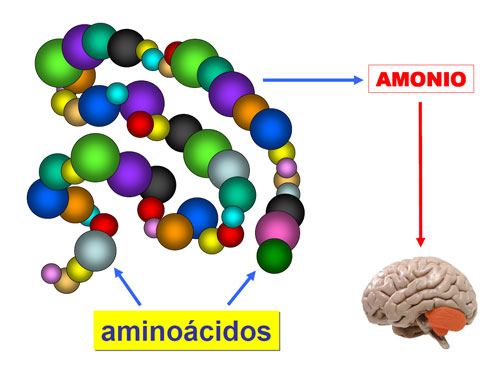
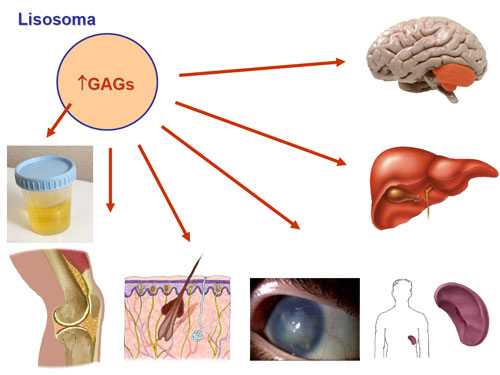
La presentación clínica varia según el grado de deficiencia enzimática, que depende, a su vez, de las mutaciones causantes del defecto de MTHFR. Las formas graves pueden manifestarse ya en el período neonatal, con hipo/hipertonía, letargia , apnea, convulsiones, etc… Las formas de presentación tardía muestran retraso mental, microcefalia , convulsiones, ataxia , trastornos psicóticos, etc… Por otra parte, la hiperhomocisteinemia es tóxica, pudiendo ocasionar complicaciones cardiovasculares ( infarto , tromboembolismo), entre otras. Además, el defecto del producto de la reacción, el MTHF, da...