¿Qué es la enfermedad de Pompe?

La enfermedad de Pompe o glucogenosis tipo II es un trastorno metabólico, causado por una acumulación de glucógeno en múltiples tejidos, principalmente el músculo, causando insuficiencia cardiaca, motora y respiratoria progresivas.
Es debida a la deficiencia de la enzima lisosomal α-glucosidasa ácida o maltasa ácida.
Fue descrita por el patólogo holandés Johannes Cassianus Pompe en 1932 en una niña de 7 meses con grave debilidad muscular, cuya autopsia mostró una masiva acumulación de glucógeno en tejidos corporales.
¿Qué es el glucógeno?
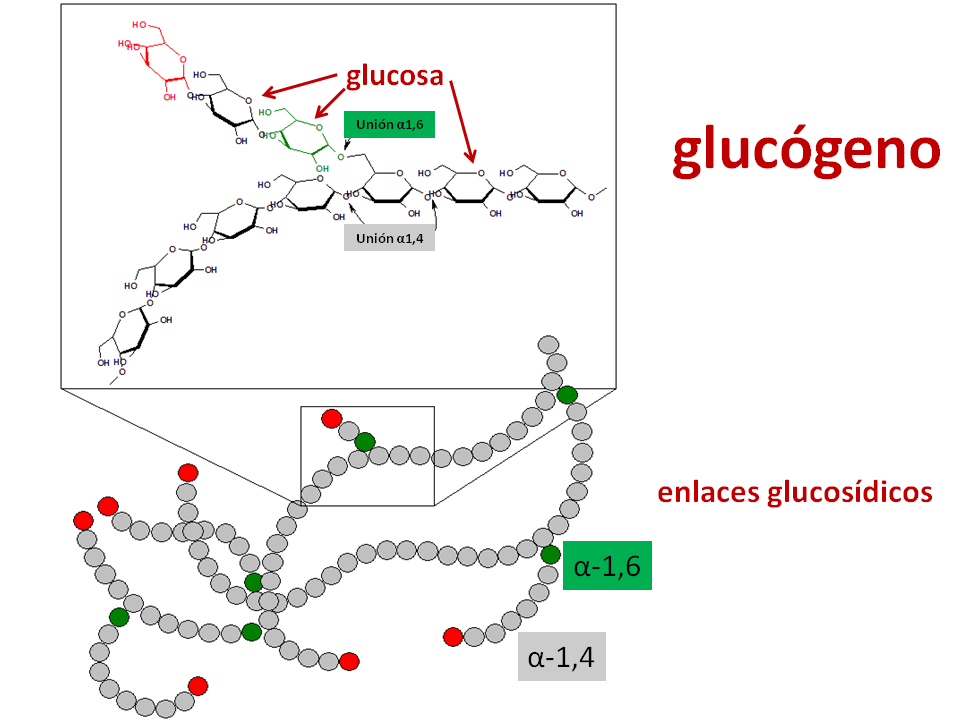
Es un polímero formado por cadenas de glucosa muy ramificadas. Su misión es liberar glucosa cuando el organismo la requiera, es decir, cuando necesite la energía que proporcionará la degradación de glucosa.
Se halla abundantemente almacenado en el hígado y, en menor cantidad, en el músculo esquelético y otros tejidos.
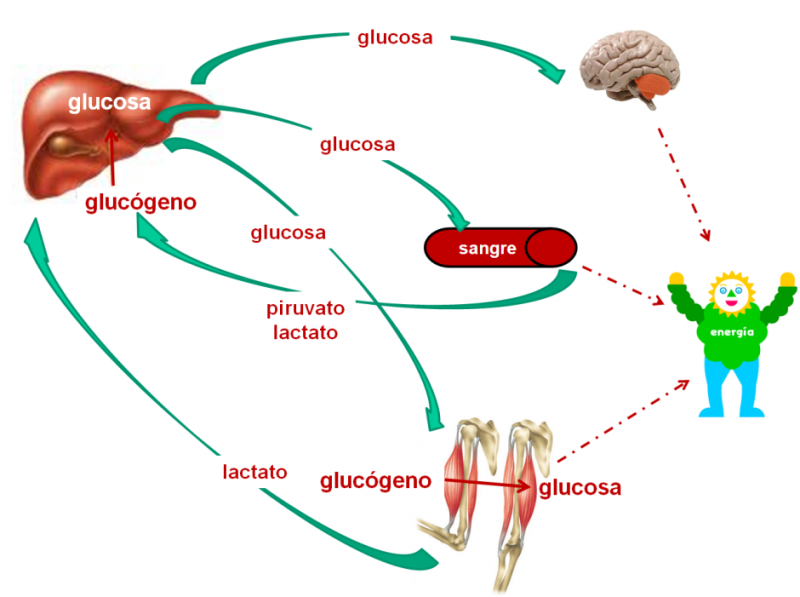
El glucógeno almacenado en el músculo, una vez convertido en glucosa, se utiliza para proporcionar a las propias fibras musculares la energía que necesitan para contraerse.
Por el contrario, el glucógeno hepático se convierte en glucosa mediante la glucogenolisis, y ésta se libera a la sangre para mantener la glucemia, siendo utilizada por todos los tejidos.
¿Qué es la α-glucosidasa ácida?
La α-glucosidasa ácida, (llamada también α-1,4-glucosidasa o maltasa ácida) es una enzima que hidroliza (rompe) el glucógeno produciendo glucosa dentro del lisosoma celular, es decir, en medio ácido, de ahí su nombre.
¿Qué es el lisosoma celular?
Es una organela o compartimento celular que contiene enzimas capaces de hidrolizar (lisar, degradar o romper) grandes moléculas, como el glucógeno, en un medio ácido.
Estas enzimas, entre ellas la α-glucosidasa ácida, se llaman hidrolasas ácidas.
¿Cómo penetra el glucógeno dentro del lisosoma celular?
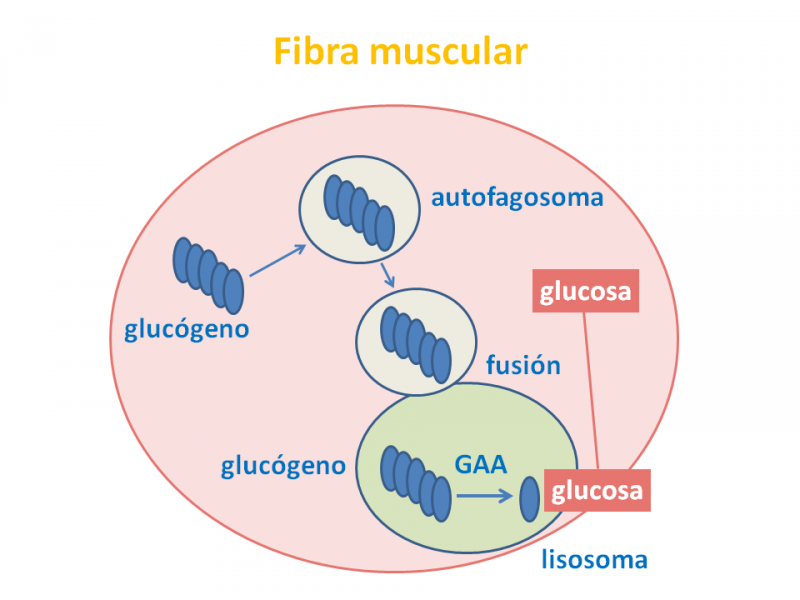
El glucógeno es una gran molécula, un polímero formado por un gran número de moléculas de glucosa (de 20.000 a 30.000), por lo que no pasa fácilmente a través de las membranas celulares.
Penetra en el lisosoma al menos en parte por autofagia, aunque se desconoce exactamente el proceso exacto.
¿Qué es la autofagia?
Es una ruta catabólica intracelular que entrega a los lisosomas proteínas envejecidas y organelas dañadas para su degradación y reciclaje.
Su función es proporcionar energía y aminoácidos para mantener la función celular en condiciones de inanición.
Además realiza una función de limpieza, liberando a la célula de proteínas mal plegadas, agregados proteicos y organelas desgastadas, que podrían interferir en los procesos metabólicos, proporcionando una especie de reciclaje, la renovación fisiológica de la célula.
Es un proceso indispensable, en el que intervienen muchas proteínas recientemente conocidas y cuya supresión es letal para la célula.
¿Por qué se produce la enfermedad de Pompe?
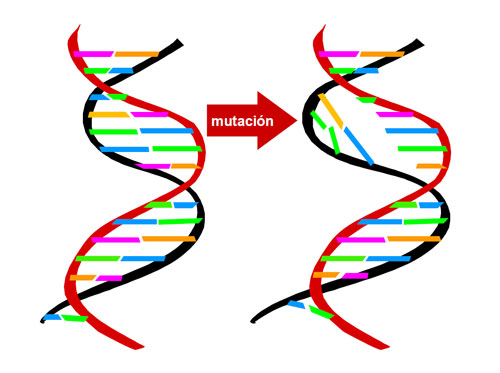
La enzima α-glucosidasa ácida está determinada genéticamente (codificada). Su deficiencia se produce por mutaciones (cambios estables y hereditarios) en el gen GAA que codifica para esta proteína enzimática.
La enfermedad de Pompe se hereda de forma autosómica recesiva, es decir, los padres son portadores de mutaciones en el gen GAA, aunque no sufren los efectos de la deficiencia de α-glucosidasa ácida.
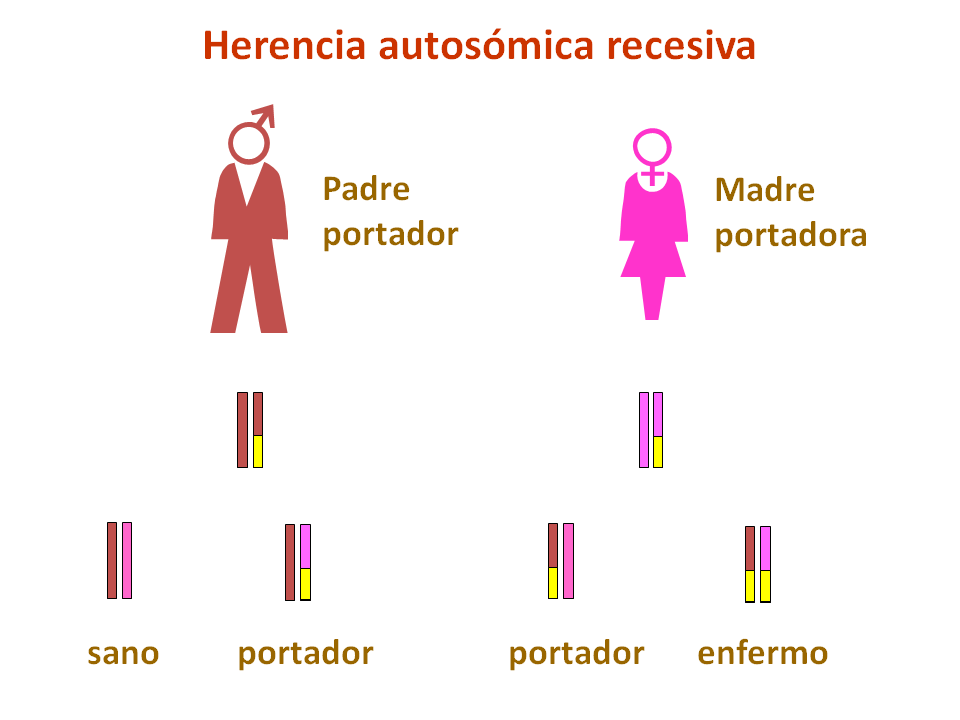
Si ambos padres transmiten una mutación en dicho gen al hijo, éste sufrirá una enfermedad de Pompe.
Consejos

Actualidad

Dieta equilibrada

Una alimentación equilibrada y adecuada es la que satisface las necesidades nutricionales de una persona, lo que supone un correcto aporte de energía y nutrientes para el buen funcionamiento del cuerpo humano.
Es importante que la alimentación sea variada para cubrir los requerimientos nutricionales y que sea agradable al paladar para evitar la monotonía de los menús.
Recursos

Todas las personas somos distintas, por dentro y por fuera. Sí, unas son morenas y tienen las piernas largas; otras son rubias y redonditas; algunas tiene orejas pequeñas y otras, los ojos como platos.
Eso es lo que podemos ver exteriormente. Pero en el interior de nuestro...

Una nueva aventura de la Pandilla Metabólica, en la que Annia, Tomás, Laura, Gabriel y Arnau tienen dos misterios para resolver.
Desde hace un tiempo, la clase de al lado saca mucho mejores notas que ellos en los exámenes. Su profesora no está nada contenta y les echa...

La Pandilla Metabólica va haciendose mayor y ya han acabado sus estudios de secundaria. Laura y Ámbar han empezado sus estudios en la Escuela de Hostelería, su gran ilusión desde pequeñas.
Pero al llegar se encuentran con que no hay ninguna asignatura especializada...

La aplicación Primeros Auxilios Fáciles proporciona, de una manera clara y sencilla, conocimientos básicos y pautas concretas para aplicar medidas de primeros auxilios en caso de urgencia antes de...

Con esta aplicación podrás pintar infinidad de mandalas y trabajar la motricidad fina y la coordinación ojo-mano, además de la creatividad y la imaginación.