Las glucogenosis musculares, origen y tipos

Son el conjunto de enfermedades hereditarias que afectan al metabolismo del glucógeno almacenado en el músculo.
En general, están originadas por deficiencias de enzimas implicadas en el metabolismo del glucógeno muscular.
Pueden causar:
- Acumulación excesiva de moléculas de glucógeno con estructura normal.
- Acumulación excesiva de moléculas de glucógeno con estructura anormal.
- Depleción de glucógeno, es decir, ausencia de síntesis del mismo.
Tienen unas características clínicas similares:
- Intolerancia al ejercicio con calambres.
- Mioglobinuria (aparición de mioglobina en orina, que es una proteína muscular y que da a la orina un color coñac).
- Debilidad progresiva, aunque su gravedad y complicaciones son diferentes.
Serán tratadas en su conjunto, a excepción de la enfermedad de Pompe (GSD-II) que se trata en su apartado correspondiente.
¿Cuáles son las principales glucogenosis musculares?

En la actualidad son un grupo de enfermedades que se identifican con números romanos, por los síndromes relacionados con la primera descripción clínica y con los nombres de las proteínas enzimáticas deficientes en cada enfermedad.
La clasificación fisiopatológica más nueva (Gazzerro E y col, Curr Neurol Neurosci Rep 2013;13:333) distingue entre:
- Glucogenosis primarias: causadas por defectos genéticos en las enzimas directamente implicadas en la síntesis de glucógeno (gluconeogenesis), la degradación (glucogenolisis) o el metabolismo de la glucosa (glucolisis).
- Glucogenosis secundarias: causadas por la pérdida de función de proteínas reguladoras que afectan indirectamente a las enzimas implicadas en las vías del glucógeno y la glucosa.
En la figura adjunta se señalan en verde las glucogenosis primordialmente musculares y en rojo las primordialmente hepáticas, aunque muchas enzimas tienen isoformas hepáticas y musculares, por lo que se manifiestan en ambos órganos (GSD III, IV, IX, 0).
¿Cómo se heredan las glucogenosis musculares?
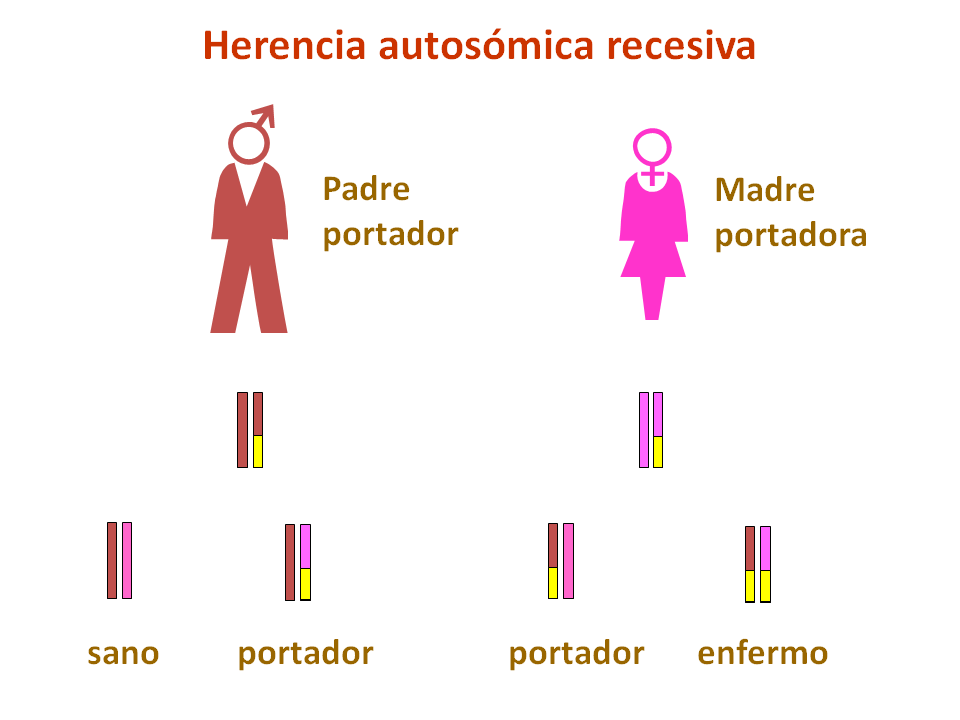
Estas deficiencias son trastornos genéticos principalmente de herencia autosómica recesiva, es decir, los padres son portadores de mutaciones en uno de estos genes aunque no sufren los efectos de la deficiencia enzimática.
Si ambos padres transmiten la mutación a niño, éste sufrirá una glucogenosis, según cuál sea la proteína mutada.
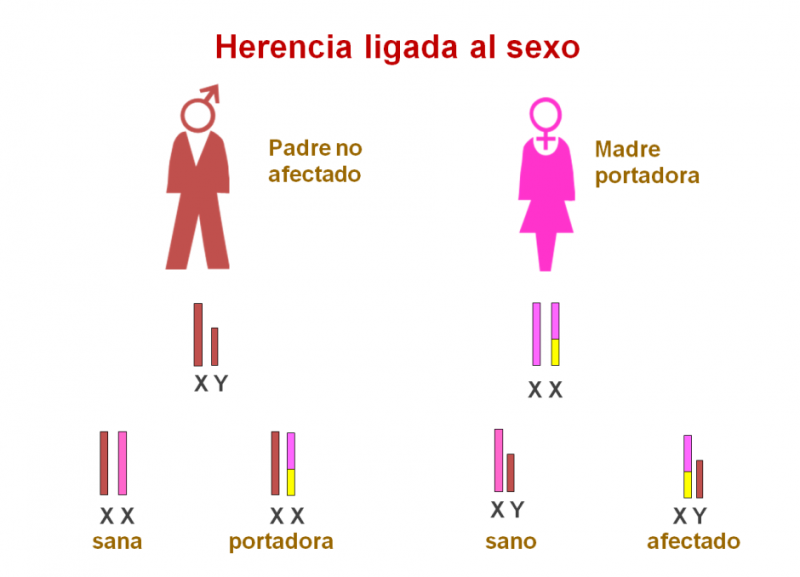
Existe alguna forma de glucogenosis muscular (GSD-VII y IX) cuyo gen está localizado en el cromosoma X, por lo que la herencia estará ligada al X, es decir, herencia materna.
 |
 |
¿Cuáles son las principales características clínicas de las glucogenosis musculares?
Las principales características clínicas de las glucogenosis musculares primarias se resumen en la tabla siguiente (modificada de Gazzerro E y col, Curr Neurol Neurosci Rep 2013;13:333).

Las GSD más frecuentes dentro de este grupo son GSD-II, GSD-III, GSD-V y GSD-VII. La GSD-II (Enfermedad de Pompe) se trata específicamente su apartado correspondiente.
La GSD-III está causada por la deficiencia de amilo 1,6-glucosidasa o enzima desramificante, cuya misión es romper los enlaces α-1,6 glucosídicos del glucógeno. Su deficiencia causa la acumulación de dextrinas, disminuyendo la liberación de glucosa.
Puede manifestarse en hígado, corazón y músculo GSD-IIIa o bien solo en hígado GSD-IIIb.
La GSD-IIIa se manifiesta en músculo en la tercera o cuarta décadas de la vida con intolerancia al ejercicio, debilidad muscular y elevación de la creatin quinasa (CK) sérica.
La afectación cardíaca es más evidente desde el punto de vista de la ecografía y variable desde el punto de vista de síntomas, pudiendo estar silente muchos años.
La GSD-V o enfermedad de McArdle está causada por la deficiencia de miofosforilasa (fosforilasa muscular). Se comienza a manifestar en la infancia, pero a menudo no se diagnostica hasta la 2ª o 3ª década de la vida.
Se presenta con intolerancia al ejercicio, mialgia (dolor muscular) y rigidez o debilidad muscular en acción que mejora con el reposo. Se tolera mal el ejercicio intenso, que causa calambres dolorosos y contracturas.
Es característico el fenómeno del 2º impulso (ver tabla anterior), que consiste en que tras una disminución de la intensidad del ejercicio, el paciente lo retoma con mayor facilidad, debido a la capacidad del cuerpo para hallar fuentes alternativas de energía, como ácidos grasos.
La GSD-VII o enfermedad de Tarui está causada por la deficiencia de la subunidad muscular de fosfofructoquinasa (PFK-M), primer paso de la glucolisis que transforma la fructosa 6-fosfato en fructosa 1,6 difosfato.
La forma más frecuente es la del adulto que se manifiesta con calambres musculares y debilidad inducidos por el ejercicio, así como mioglobinuria e hiperbilirrubinemia (aumento de la bilirrubina en sangre), con hemólisis (ruptura de glóbulos rojos) compensada.
Existe una forma infantil menos frecuente, con debilidad generalizada y afectación de otros órganos: convulsiones, ceguera cortical, opacidades corneales y cardiomiopatía.
Las principales características clínicas de las glucogenosis musculares causantes de depleción de glucógeno y secundarias se resumen en la tabla siguiente (Gazzerro E y col, Curr Neurol Neurosci Rep 2013;13:333).

Consejos

Actualidad

Dieta equilibrada

Una alimentación equilibrada y adecuada es la que satisface las necesidades nutricionales de una persona, lo que supone un correcto aporte de energía y nutrientes para el buen funcionamiento del cuerpo humano.
Es importante que la alimentación sea variada para cubrir los requerimientos nutricionales y que sea agradable al paladar para evitar la monotonía de los menús.
Recursos

Todas las personas somos distintas, por dentro y por fuera. Sí, unas son morenas y tienen las piernas largas; otras son rubias y redonditas; algunas tiene orejas pequeñas y otras, los ojos como platos.
Eso es lo que podemos ver exteriormente. Pero en el interior de nuestro...

Una nueva aventura de la Pandilla Metabólica, en la que Annia, Tomás, Laura, Gabriel y Arnau tienen dos misterios para resolver.
Desde hace un tiempo, la clase de al lado saca mucho mejores notas que ellos en los exámenes. Su profesora no está nada contenta y les echa...

La Pandilla Metabólica va haciendose mayor y ya han acabado sus estudios de secundaria. Laura y Ámbar han empezado sus estudios en la Escuela de Hostelería, su gran ilusión desde pequeñas.
Pero al llegar se encuentran con que no hay ninguna asignatura especializada...

La aplicación Primeros Auxilios Fáciles proporciona, de una manera clara y sencilla, conocimientos básicos y pautas concretas para aplicar medidas de primeros auxilios en caso de urgencia antes de...

Con esta aplicación podrás pintar infinidad de mandalas y trabajar la motricidad fina y la coordinación ojo-mano, además de la creatividad y la imaginación.