Las glucogenosis hepáticas, origen y tipos

Las glucogenosis hepáticas son el conjunto de enfermedades hereditarias que afectan al metabolismo del glucógeno almacenado en el hígado. En general, están causadas por deficiencias de enzimas implicadas en el metabolismo hepático del glucógeno.
Las GSD hepáticas serán tratadas en su conjunto, porque tienen unas características clínicas similares (hepatomegalia, hipoglucemia y retraso del crecimiento), aunque su gravedad y complicaciones son diferentes.
¿Cuáles son las principales glucogenosis hepáticas?
Se denominan por números romanos, por los síndromes relacionados con la primera descripción clínica y con los nombres de las proteínas enzimáticas deficientes en cada enfermedad.
Estos nombres, así como los órganos primordialmente afectados en ellas están resumidos en la siguiente tabla.
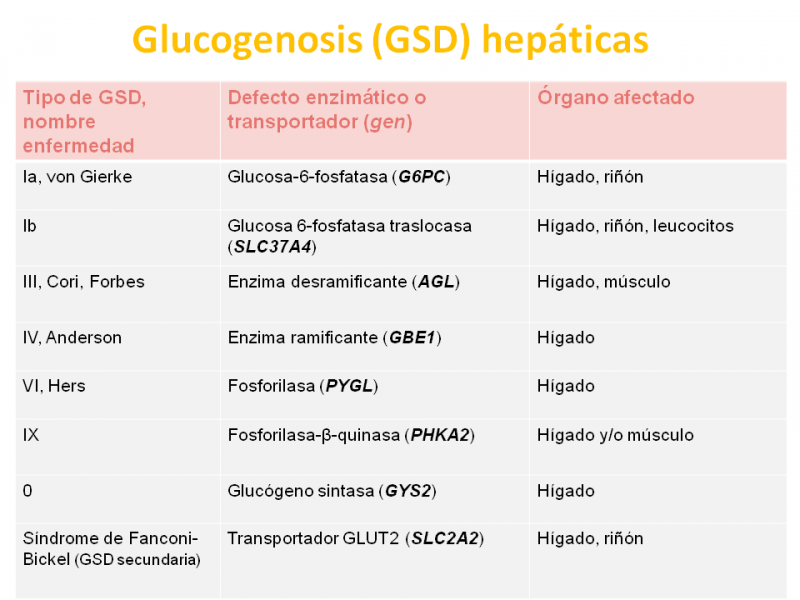
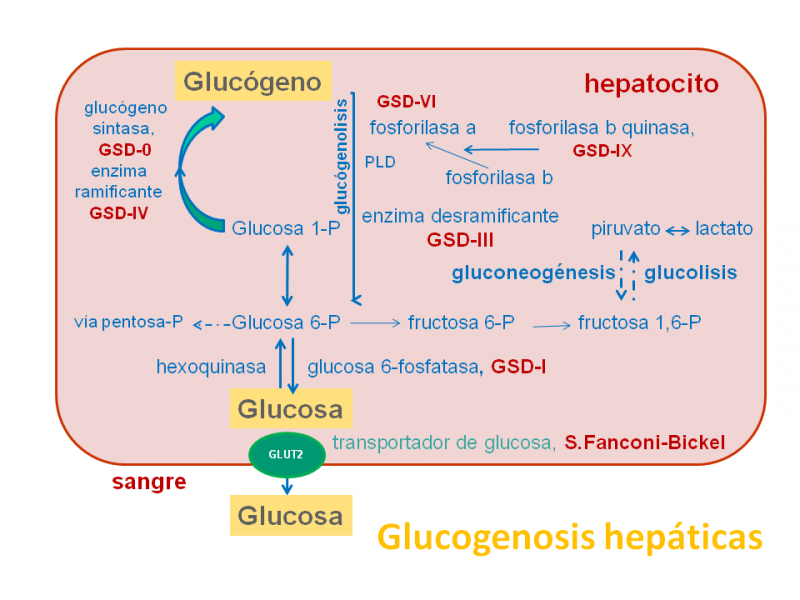
En la siguiente figura se señalan las glucogenosis (GSD) hepáticas en color rojo.
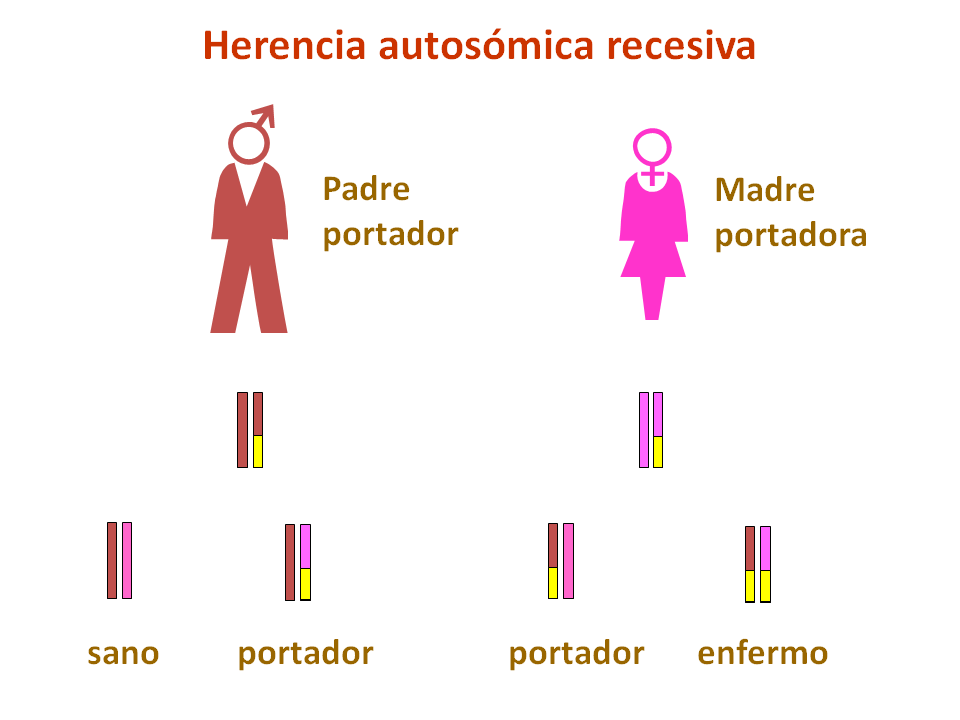
¿Cómo se heredan las glucogenosis hepáticas?
Estas deficiencias son trastornos genéticos principalmente de herencia autosómica recesiva, es decir, los padres son portadores de mutaciones en uno de estos genes, aunque no sufren los efectos de la deficiencia enzimática. Si ambos padres transmiten la mutación al niño, éste sufrirá una glucogenosis.
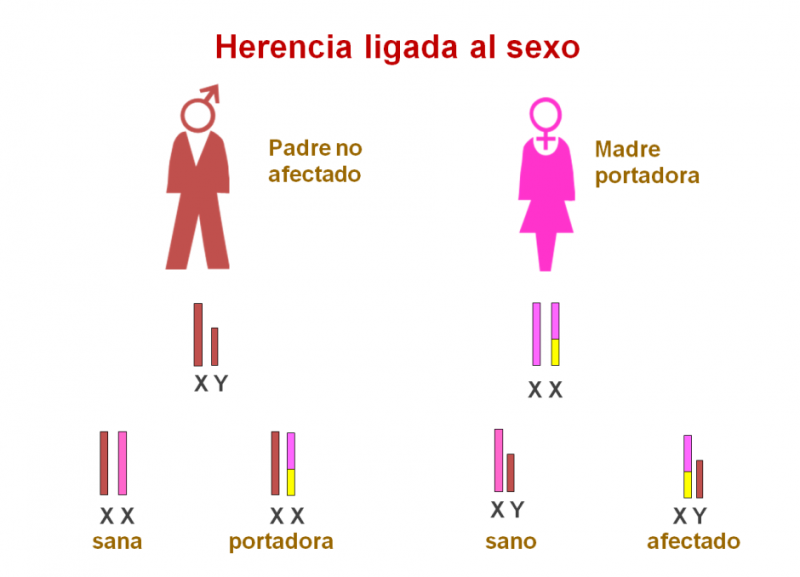
Existe una forma de glucogenosis (GSD-IXa) ligada al cromosoma X, es decir, de herencia materna.
 |
 |
¿Cuáles son las principales características clínicas y bioquímicas de las glucogenosis hepáticas?
Todas las glucogenosis hepáticas se caracterizan clínicamente por presentar hepatomegalia y retraso del crecimiento, pero la afectación hepática es muy grave en la GSD- Ia, causada por la deficiencia de glucosa 6-fosfatasa, ya que dicha deficiencia impide la formación de glucosa a partir de glucosa 6-P.
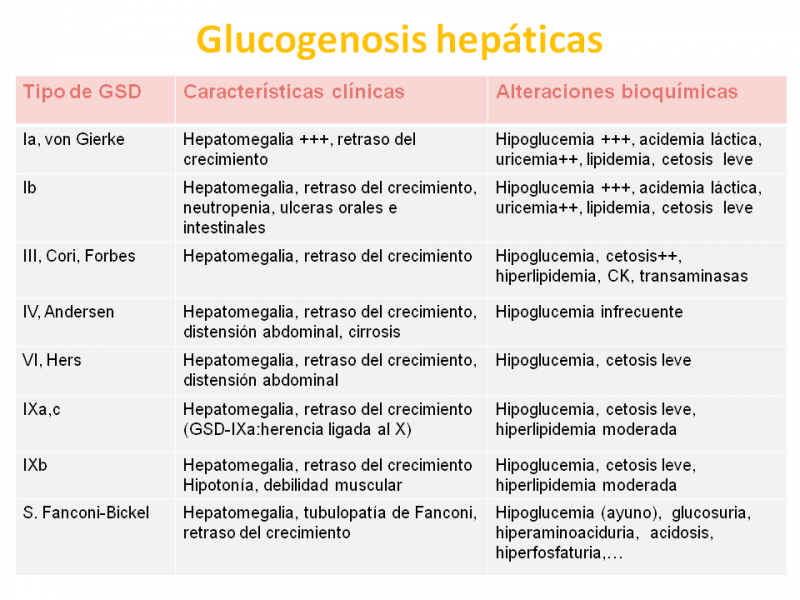
La hepatopatía puede aparecer ya en el nacimiento o en el período neonatal y va acompañada de hipoglucemia grave, acidemia láctica, acidosis metabólica, hiperuricemia e hiperlipidemia.
La variante GSD-Ib está causada por la deficiencia de la traslocasa de glucosa 6-fosfato al retículo endoplasmático, donde la glucosa 6-fosfatasa transforma la glucosa 6-P en glucosa, que posteriormente pasa a la sangre.
La GSD-Ib, que tiene el mismo efecto bioquímico que la GSD-Ia, se presenta además con neutropenia (disminución del número de neutrófilos en sangre) desde leve hasta agranulocitosis (disminución importante del número de neutrófilos), que determina la aparición de úlceras orales o intestinales.
La GSD-III está causada por la deficiencia de amilo 1,6-glucosidasa o enzima desramificante (rompe los enlaces α-1,6 glucosídicos). Existen variantes más frecuentes de expresión hepática y muscular (IIIa) o solo hepática (IIIb). La expresión clínica es más leve que la GSD-I y toleran mejor el ayuno.
La GSD-IV o amilopectinosis está causada por la deficiencia de enzima ramificante, que causa la acumulación de un glucógeno anómalo, no ramificado, parecido a la amilopectina (poliglucosán: polímero de glucosa) que se acumula en tejidos causando daño celular.
La GSD-IV se presenta entre los 3-15 meses con hepatomegalia, fallo de medro y distensión abdominal, y puede derivar a una hepatopatía crónica (cirrosis). Existen formas de afectación multisistémica, que afectan hígado y músculo, presentando además hipotonía y miocardiopatía.
Los defectos del sistema de la fosforilasa hepática comprenden las deficiencias de fosforilasa hepática (GSD VI) y de fosforilasa quinasa (GDS-IX).
La fosforilasa hepática rompe las cadenas lineales de glucógeno para producir glucosa 1-P, pero debe activarse mediante la acción de la fosforilasa quinasa, es decir, que actúan coordinadamente.
Las formas hepáticas de ambos tipos tienen un cuadro clínico similar, más leve que los anteriores y con mejor pronóstico.La GSD-IX tiene 6 subtipos, siendo los hepáticos las formas GSD IXa (de herencia ligada al cromosoma X y la más frecuente) y GSD IXc (no ligada al X) y la GSD-IXb, hepática y muscular.
La deficiencia de la isoforma hepática de glucógeno sintetasa o GSD-0, está causada por un defecto en la capacidad de acumular glucógeno y no presenta hepatomegalia, aunque sí talla baja e hipoglucemia cetósica.
Finalmente, la deficiencia de GLUT2, aunque es una alteración de transporte de glucosa, los pacientes muestran una clínica similar a las GSD, con alteración de la glucogenolisis y neoglucogénesis (formación de glucosa a partir del piruvato).
Las manifestaciones clínicas aparecen entre los 3-10 meses y consisten en hepatomegalia, tubulopatía de Fanconi (glucosuria, hiperfosfaturia, hiperaminoaciduria).
Consejos

Actualidad

Dieta equilibrada

Una alimentación equilibrada y adecuada es la que satisface las necesidades nutricionales de una persona, lo que supone un correcto aporte de energía y nutrientes para el buen funcionamiento del cuerpo humano.
Es importante que la alimentación sea variada para cubrir los requerimientos nutricionales y que sea agradable al paladar para evitar la monotonía de los menús.
Recursos

Todas las personas somos distintas, por dentro y por fuera. Sí, unas son morenas y tienen las piernas largas; otras son rubias y redonditas; algunas tiene orejas pequeñas y otras, los ojos como platos.
Eso es lo que podemos ver exteriormente. Pero en el interior de nuestro...

Una nueva aventura de la Pandilla Metabólica, en la que Annia, Tomás, Laura, Gabriel y Arnau tienen dos misterios para resolver.
Desde hace un tiempo, la clase de al lado saca mucho mejores notas que ellos en los exámenes. Su profesora no está nada contenta y les echa...

La Pandilla Metabólica va haciendose mayor y ya han acabado sus estudios de secundaria. Laura y Ámbar han empezado sus estudios en la Escuela de Hostelería, su gran ilusión desde pequeñas.
Pero al llegar se encuentran con que no hay ninguna asignatura especializada...

La aplicación Primeros Auxilios Fáciles proporciona, de una manera clara y sencilla, conocimientos básicos y pautas concretas para aplicar medidas de primeros auxilios en caso de urgencia antes de...

Con esta aplicación podrás pintar infinidad de mandalas y trabajar la motricidad fina y la coordinación ojo-mano, además de la creatividad y la imaginación.