¿Qué es la fenilcetonuria?
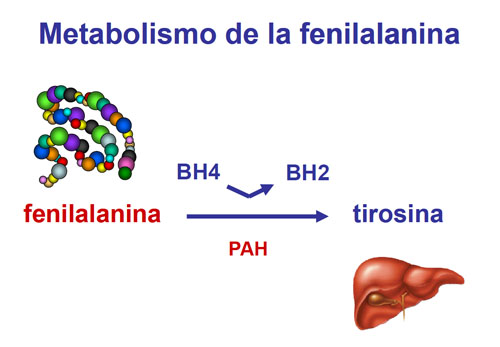
La fenilcetonuria (PKU) es un error congénito del metabolismo, en el que la deficiencia de la enzima fenilalanina hidroxilasa, causa una acumulación de fenilalanina en sangre, orina y tejidos, así como un defecto de tirosina.
¿Qué es la fenilalanina?
La fenilalanina es un aminoácido, molécula simple que forma parte de las proteínas.
Las proteínas están constituidas por una cadena muy larga de aminoácidos, que se enlazan como las perlas de un collar, en un orden especial para cada una de ellas, que determina su forma en el espacio y con ello, su buen funcionamiento.
Cuando las proteínas se degradan, se liberan los aminoácidos y estos pueden utilizarse para formar otras proteínas nuevas de nuestro organismo o bien para generar otras sustancias y energía.
La fenilalanina tiene su propia vía metabólica, por la cual es capaz de formar un aminoácido muy parecido a ella, la tirosina, gracias a la acción de una enzima, la fenilalanina hidroxilasa (PAH) y de un coenzima que facilita la reacción, la tetrahidrobiopterina (BH4).
Consejos

Actualidad

Nutrición

Recursos

Todas las personas somos distintas, por dentro y por fuera. Sí, unas son morenas y tienen las piernas largas; otras son rubias y redonditas; algunas tiene orejas pequeñas y otras, los ojos como platos.
Eso es lo que podemos ver exteriormente. Pero en el interior de nuestro...


Una nueva aventura de la Pandilla Metabólica, en la que Annia, Tomás, Laura, Gabriel y Arnau tienen dos misterios para resolver.
Desde hace un tiempo, la clase de al lado saca mucho mejores notas que ellos en los exámenes. Su profesora no está nada contenta y les echa...

El mundo de los trastornos metabólicos está lleno de palabras complicadas, como fenilalanina, tirosinemia,...

La Pandilla Metabólica va haciendose mayor y ya han acabado sus estudios de secundaria. Laura y Ámbar han empezado sus estudios en la Escuela de Hostelería, su gran ilusión desde pequeñas.
Pero al llegar se encuentran con que no hay ninguna asignatura especializada...