¿Qué ocurre en la PKU?

Cuando existe un error en el metabolismo de la fenilalanina, ésta no puede convertirse fácilmente en tirosina porque falla la enzima que interviene en esta reacción, la PAH.
Esto causa una acumulación de la fenilalanina en sangre, orina, tejidos y en el cerebro.
Además de la fenilalanina se acumulan también unos compuestos que se forman a partir de ella, las fenilcetonas, que se eliminan por la orina y son las que dan el nombre a la enfermedad: fenilcetonuria o PKU (del inglés Phenyl-Keton-Uria).
Otra consecuencia de este defecto es la falta de síntesis de tirosina, aminoácido muy importante como precursor de neurotransmisores.
¿Por qué se produce una deficiencia de actividad de la PAH?
La deficiencia de actividad PAH se produce debido a mutaciones (cambios estables y hereditarios) en el gen PAH que codifica a esta enzima.
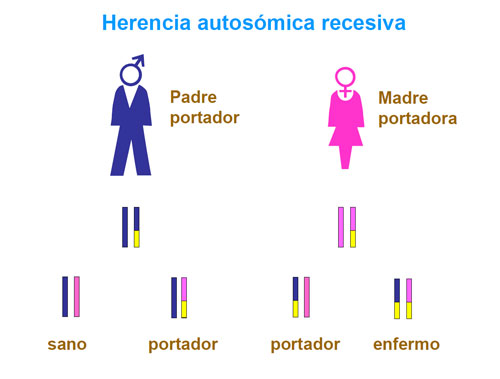
La PKU es un trastorno genético de herencia autosómica recesiva, es decir, los padres son portadores de mutaciones en el gen PAH, aunque no sufran los efectos de la deficiencia enzimática.
Si ambos padres transmiten una mutación al hijo, dependiendo de la severidad de las mismas, el niño mostrará un defecto de actividad enzimática total o parcial, que dará lugar a una PKU o a una hiperfenilalaninemia moderada, respectivamente.
Consejos

Actualidad

Nutrición

Recursos

Todas las personas somos distintas, por dentro y por fuera. Sí, unas son morenas y tienen las piernas largas; otras son rubias y redonditas; algunas tiene orejas pequeñas y otras, los ojos como platos.
Eso es lo que podemos ver exteriormente. Pero en el interior de nuestro...


Una nueva aventura de la Pandilla Metabólica, en la que Annia, Tomás, Laura, Gabriel y Arnau tienen dos misterios para resolver.
Desde hace un tiempo, la clase de al lado saca mucho mejores notas que ellos en los exámenes. Su profesora no está nada contenta y les echa...

El mundo de los trastornos metabólicos está lleno de palabras complicadas, como fenilalanina, tirosinemia,...

La Pandilla Metabólica va haciendose mayor y ya han acabado sus estudios de secundaria. Laura y Ámbar han empezado sus estudios en la Escuela de Hostelería, su gran ilusión desde pequeñas.
Pero al llegar se encuentran con que no hay ninguna asignatura especializada...