¿Cómo se diagnostica la PKU?
En la mayor parte de países existe un programa de detección precoz que permite detectar los recién nacidos que la padecen en los primeros días de la vida, con objeto de aplicar un tratamiento precoz, antes de que se manifiesten los síntomas de la enfermedad.
¿Qué es la detección precoz neonatal?
Es un programa de salud pública destinado a identificar entre todos los neonatos de una población, a aquellos con mayor riesgo de sufrir determinadas enfermedades: genéticas (Errores Congénitos del Metabolismo, como la Fenilcetonuria), endocrinas, embrionarias o infecciosas.
¿Detección precoz es igual a diagnóstico?
La detección precoz es un paso previo al diagnóstico, pues permite identificar entre todos los neonatos de un país, a aquellos que tienen mayor probabilidad de padecer una determinada enfermedad, como la fenilcetonuria.
Estos neonatos, una vez detectados, requieren confirmación y diagnóstico clínico, bioquímico y, en algunos casos, genético.
¿Cuál es el objetivo de la detección precoz?
El identificar en el período neonatal (precozmente) alguna de estas enfermedades permite un tratamiento precoz de las mismas, antes de que se hayan instaurado las manifestaciones clínicas.
Una vez manifestados los problemas, básicamente neurológicos (en el caso de la fenilcetonuria), las opciones terapéuticas son escasas.
¿Cuándo empezó la detección precoz de la fenilcetonuria?
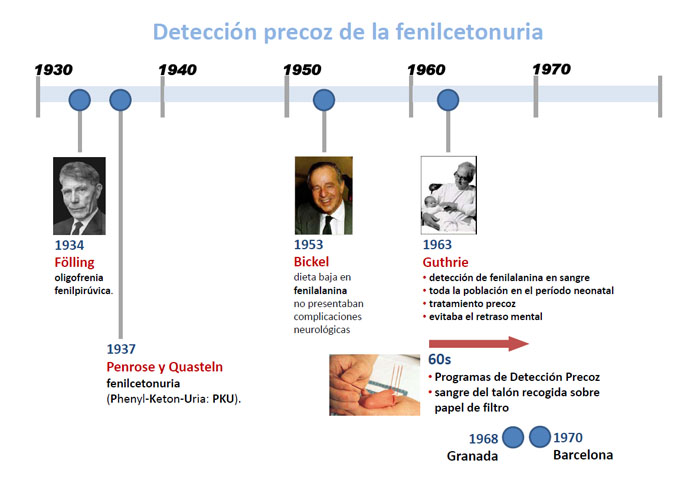
- En 1934 Fölling descubrió un trastorno metabólico que cursaba con retraso mental, al que denominó oligofrenia fenilpirúvica.
- En 1937 Penrose y Quastel la denominaron fenilcetonuria por la elevada excreción de fenilcetonas (Phenyl-Keton-Uria: PKU).
- En 1953, Bickel demostró que los pacientes con fenilcetonuria tratados con una dieta baja en fenilalanina no presentaban las complicaciones neurológicas propias de la enfermedad.
- En 1963 Guthrie ideó un método de detección de fenilalanina en sangre para ser aplicado a toda la población en el período neonatal, lo que permitía el tratamiento precoz de la PKU y evitaba el retraso mental de los pacientes tratados prematuramente.
- A partir de los años 60 aparecieron los primeros Programas de Detección Precoz de la enfermedad mediante la determinación de fenilalanina en la muestra de sangre del talón recogida sobre papel de filtro.
¿Cuándo se inició la detección precoz en España?
La detección precoz de la fenilcetonuria y el hipotiroidismo congénito se inició a partir del año 1968 en Granada.
Posteriormente comenzaron otros programas similares en Barcelona (1970) y Madrid (1973).
El Plan Nacional para la Prevención de la Subnormalidad elaborado bajo los auspicios del Real Patronato para la Discapacidad fue el impulsor de la expansión de los programas de detección en toda España.
¿Cuál es actualmente la cobertura del programa?
La cobertura del programa es del 99,5%, prácticamente total para todo el territorio español.
¿Cuál es la incidencia de hiperfenilalaninemia en Cataluña?
La incidencia global de casos positivos en los últimos 27 años en Cataluña fue de 1:10105 recién nacidos vivos (PKU 1:18.636 e hiperfenilalaninemia moderada 1:22072).
¿Cuál es el paso siguiente una vez detectado un nuevo caso?
Cuando el resultado del cribaje metabólico es positivo se avisa inmediatamente a la familia indicándole que acuda a la Unidad de tratamiento y seguimiento correspondiente.
El centro de referencia aplica un protocolo:
- Confirmación del diagnóstico.
- Diagnóstico diferencial.
- Tratamiento.
- Seguimiento del paciente con PKU.
¿En qué consiste la confirmación del diagnóstico?
En una determinación de fenilalanina en plasma del bebé que permite confirmar que la elevación de este aminoácido observada en el Centro de detección precoz no es transitoria.
¿Y el diagnóstico diferencial… qué es?
Existen varias causas secundarias de elevación plasmática de fenilalanina (defectos del metabolismo de pterinas), pero la más importante es la fenilcetonuria.
En la sangre extraída al bebé y en una muestra de orina se pueden excluir estas causas secundarias. El estudio genético confirmará definitivamente el diagnóstico.
¿Cuándo se instaura el tratamiento?
Inmediatamente después del diagnóstico (ver tratamiento de la PKU).
¿Cuánto tiempo dura el seguimiento?
La PKU es una enfermedad genética con buen pronóstico si se trata precozmente y el control metabólico es bueno, por lo que el seguimiento dura toda la vida.
Consejos

Actualidad

Nutrición

Recursos

Todas las personas somos distintas, por dentro y por fuera. Sí, unas son morenas y tienen las piernas largas; otras son rubias y redonditas; algunas tiene orejas pequeñas y otras, los ojos como platos.
Eso es lo que podemos ver exteriormente. Pero en el interior de nuestro...


Una nueva aventura de la Pandilla Metabólica, en la que Annia, Tomás, Laura, Gabriel y Arnau tienen dos misterios para resolver.
Desde hace un tiempo, la clase de al lado saca mucho mejores notas que ellos en los exámenes. Su profesora no está nada contenta y les echa...

El mundo de los trastornos metabólicos está lleno de palabras complicadas, como fenilalanina, tirosinemia,...

La Pandilla Metabólica va haciendose mayor y ya han acabado sus estudios de secundaria. Laura y Ámbar han empezado sus estudios en la Escuela de Hostelería, su gran ilusión desde pequeñas.
Pero al llegar se encuentran con que no hay ninguna asignatura especializada...