¿Qué es la Alcaptonuria?
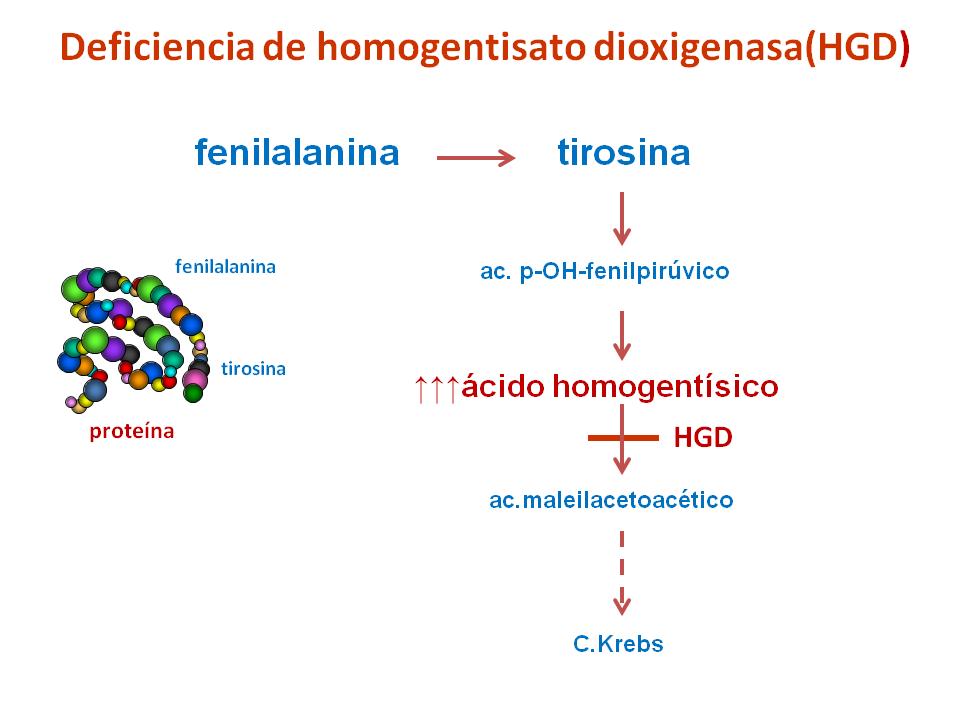
La alcaptonuria es un error congénito del metabolismo de los aminoácidos fenilalanina y tirosina, causado por la deficiencia de la enzima homogentisato dioxigenasa (HGD), que determina la acumulación de ácido homogentísico en sangre y orina.
Se caracteriza por la orina de color oscuro, ocronosis (pigmentación del tejido conjuntivo) y artrosis degenerativa de las articulaciones.
¿Qué es el ácido homogentísico?
Es un compuesto intermedio de la vía de degradación de unos aminoácidos (fenilalanina y tirosina) hacia el ciclo de Krebs. Es el sustrato de la enzima HGD y, en condiciones normales, es prácticamente indetectable en sangre y orina.
¿Cuándo se acumula el ácido homogentísico?
Cuando dicha vía catabólica (de degradación) está interferida por una deficiencia enzimática de HGD, el ácido homogentísico se acumula en sangre y se elimina en grandes cantidades en la orina.
En contacto con el aire el ácido homogentísico se oxida y polimeriza, dando lugar al pigmento negro alcaptón, que da color a la orina de los individuos que padecen esta enfermedad, a la que da el nombre (alcapton-uria).
El pigmento se deposita también en tejido conjuntivo (ocronosis), dándole un aspecto grisáceo y causando su degeneración.
Cuando se deposita en las articulaciones causa una artropatía degenerativa dolorosa y discapacitante.
¿Por qué se produce un defecto de la enzima HGD?
La deficiencia de HGD se produce debido a mutaciones (cambios estables y hereditarios) en el gen que codifica esta proteína enzimática, el gen HGO.
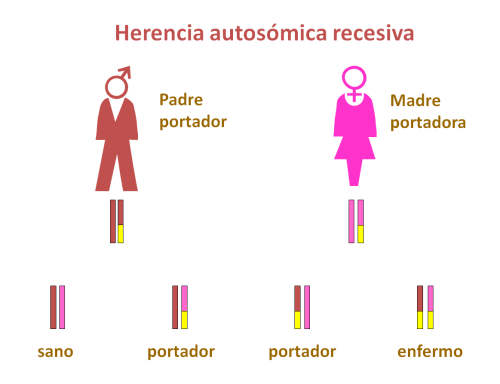
La alcaptonuria se transmite de forma autosómica recesiva, es decir, ambos padres son portadores de una mutación en el gen HGO, aunque no padecen ninguna manifestación clínica por ello.
Si ambos padres pasan al hijo un alelo mutado de este gen, el niño sufrirá una alcaptonuria.
Consejos

Actualidad

Nutrición

Recursos

Todas las personas somos distintas, por dentro y por fuera. Sí, unas son morenas y tienen las piernas largas; otras son rubias y redonditas; algunas tiene orejas pequeñas y otras, los ojos como platos.
Eso es lo que podemos ver exteriormente. Pero en el interior de nuestro...

Una nueva aventura de la Pandilla Metabólica, en la que Annia, Tomás, Laura, Gabriel y Arnau tienen dos misterios para resolver.
Desde hace un tiempo, la clase de al lado saca mucho mejores notas que ellos en los exámenes. Su profesora no está nada contenta y les echa...

La Pandilla Metabólica va haciendose mayor y ya han acabado sus estudios de secundaria. Laura y Ámbar han empezado sus estudios en la Escuela de Hostelería, su gran ilusión desde pequeñas.
Pero al llegar se encuentran con que no hay ninguna asignatura especializada...

La aplicación Primeros Auxilios Fáciles proporciona, de una manera clara y sencilla, conocimientos básicos y pautas concretas para aplicar medidas de primeros auxilios en caso de urgencia antes de...

Con esta aplicación podrás pintar infinidad de mandalas y trabajar la motricidad fina y la coordinación ojo-mano, además de la creatividad y la imaginación.