¿Qué son los defectos de la tetrahidrobiopterina (BH4)?
. Imagen: HSJDBCN")
Son un conjunto de enfermedades causadas por mutaciones en los genes implicados en las vías de síntesis o reciclaje de la BH4.
La alteración de las proteínas enzimáticas mutadas causa un defecto de BH4 que a su vez, da lugar a las deficiencias de actividad de las enzimas en las que la BH4 actúa como cofactor esencial: fenilalanina hidroxilasa, tirosina hidroxilasa y triptófano hidroxilasa y tres isoformas de óxido nítrico sintasa.
Causan un defecto en la síntesis de neurotransmisores (L-Dopa y serotonina) y pueden o no ir acompañados de hiperfenilalaninemia.
¿Qué es la BH4?
Es una pterina que actúa como cofactor esencial en diversas reacciones enzimáticas de gran importancia metabólica, como la hidroxilación de aminoácidos aromáticos (fenilalanina, tirosina y triptófano), así como en las tres isoformas de óxido nítrico sintasa.
La fenilalanina hidroxilasa (PAH) es la enzima que transforma la fenilalanina en tirosina y su defecto genético causa la fenilcetonuria (PKU).
La tirosina hidroxilasa (TH) está implicada en la síntesis de L-Dopa a partir de la tirosina y su defecto genético causa una deficiencia de este neurotransmisor.
La triptófano hidroxilasa (TPH) está implicada en la síntesis de serotonina a partir del triptófano y su defecto genético causa una deficiencia de este neurotransmisor.
Las óxido nítrico sintasas (NOS) son enzimas implicadas en la síntesis del óxido nítrico a partir de la arginina e intervienen en la comunicación celular (isoforma neuronal), la función endotelial (isoforma endotelial) y la defensa inmune y sistema cardiovascular isoforma inducible), entre otras funciones.
¿Cómo se metaboliza la BH4?
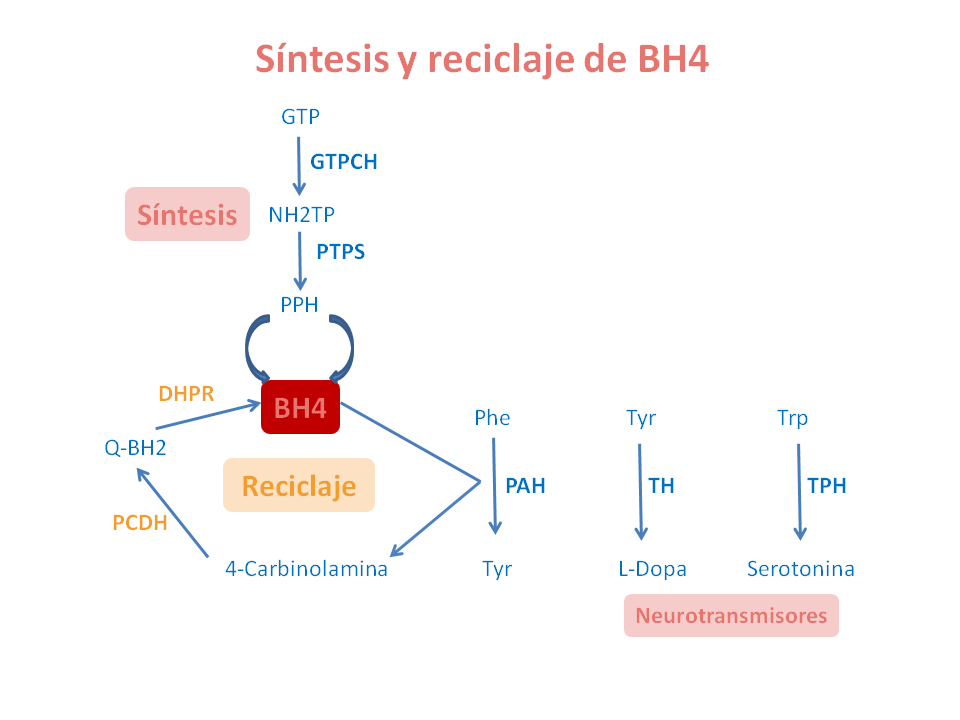
La BH4 se sintetiza a partir de guanosina trifosfato (GTP) por la acción de tres enzimas, GTP ciclohidrolasa (GTPCH), 6-piruvoil-tetrahidrobiopterina sintasa (PTPS) y sepiapterina reductasa (SR).
Como la síntesis de BH4 no es suficiente para cubrir sus importantes funciones, el cofactor debe regenerarse, lo que hace mediante dos enzimas: pterina-4-carbinolamina deshidratasa (PCD) y dihidropterina reductasa (DHPR).
¿Cuáles son los defectos del metabolismo de BH4?
Los defectos de BH4 están causados por la actividad deficiente de las enzimas implicadas en la síntesis (GTPCH, PTPS y SR) y el reciclaje (PCD y DHPR) de BH4.
¿Por qué se producen defectos en el metabolismo de BH4?
Cada una de las reacciones del metabolismo que van a dar lugar a los compuestos que forman nuestro cuerpo está determinada genéticamente (codificada). Todos heredamos de nuestros padres la información correcta o alterada que determina que se realice cada uno de estos procesos del metabolismo.
Los defectos de BH4 se producen debido a mutaciones (cambios estables y hereditarios) en los genes (GTPCH, PTPS, SR, PCD y DHPR) que codifican estas proteínas enzimáticas.
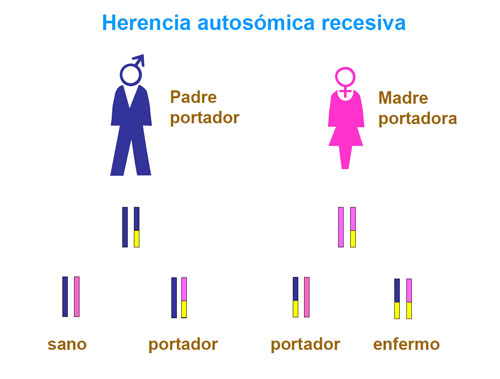
Estos defectos son trastornos genéticos de herencia autosómica recesiva, es decir, los padres suelen ser portadores de mutaciones en estos genes aunque no sufren los efectos de la deficiencia enzimática.
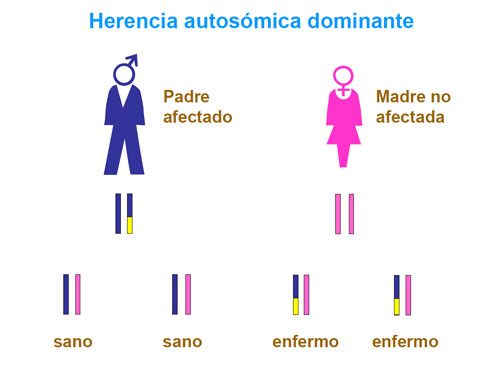
Si ambos padres transmiten al hijo un gen mutado, el niño/a sufrirá un defecto de BH4. No obstante, la GTPCH puede presentar también una herencia dominante, como en la enfermedad de Segawa, que es la más frecuente.
 |
 |
Consejos

Actualidad

Dieta equilibrada

Una alimentación equilibrada y adecuada es la que satisface las necesidades nutricionales de una persona, lo que supone un correcto aporte de energía y nutrientes para el buen funcionamiento del cuerpo humano.
Es importante que la alimentación sea variada para cubrir los requerimientos nutricionales y que sea agradable al paladar para evitar la monotonía de los menús.
Recursos

Todas las personas somos distintas, por dentro y por fuera. Sí, unas son morenas y tienen las piernas largas; otras son rubias y redonditas; algunas tiene orejas pequeñas y otras, los ojos como platos.
Eso es lo que podemos ver exteriormente. Pero en el interior de nuestro...

Una nueva aventura de la Pandilla Metabólica, en la que Annia, Tomás, Laura, Gabriel y Arnau tienen dos misterios para resolver.
Desde hace un tiempo, la clase de al lado saca mucho mejores notas que ellos en los exámenes. Su profesora no está nada contenta y les echa...

La Pandilla Metabólica va haciendose mayor y ya han acabado sus estudios de secundaria. Laura y Ámbar han empezado sus estudios en la Escuela de Hostelería, su gran ilusión desde pequeñas.
Pero al llegar se encuentran con que no hay ninguna asignatura especializada...

La aplicación Primeros Auxilios Fáciles proporciona, de una manera clara y sencilla, conocimientos básicos y pautas concretas para aplicar medidas de primeros auxilios en caso de urgencia antes de...

Con esta aplicación podrás pintar infinidad de mandalas y trabajar la motricidad fina y la coordinación ojo-mano, además de la creatividad y la imaginación.