Defectos del metabolismo de los aminoácidos ramificados (II): otros defectos metabólicos comunes a los 3 AAR menos frecuentes
: otros defectos metabólicos comunes a los 3 AAR menos frecuentes")
La Dra. Àngels García-Carzola, jefa de la Unidad de Enfermedades Metabólicas del Hospital Sant Joan de Déu Barcelona, impartió la conferencia sobre los defectos del metabolismo de los aminoácidos ramificados en el simposio anual SSIEM (septiembre de 2019), que reúne a expertos y profesionales de todo el mundo relacionados con los errores congénitos del metabolismo.
En la primera parte de este resumen se comentaba el defecto metabólico más frecuente de la vía de los aminoácidos ramificados que da lugar a la enfermedad de jarabe de arce. A continuación, se comentarán otros defectos mucho menos frecuentes, de reciente descripción, con una presentación clínica más leve y potencialmente tratables.
Deficiencias de la aminotransferasa de los AAR
La deficiencia de la aminotransferasa mitocondrial de los AAR (BCAT2), primera enzima de la degradación de dichos aminoácidos. Se han descrito mutaciones en el gen BCAT2 en seis pacientes con elevada concentración de los AAR, pero no de sus cetoácidos ni de aloisoleucina a diferencia de los pacientes con MSUD.
Los síntomas clínicos (a excepción de la ausencia de encefalopatía) son diversos: dos de ellos eran asintomáticos, mientras que tres presentaban un retraso del desarrollo, trastorno dentro del espectro autista y, uno de ellos, ataxia (Knerr y col, 2019, Temple Street Children's University Hospital, Dublin).
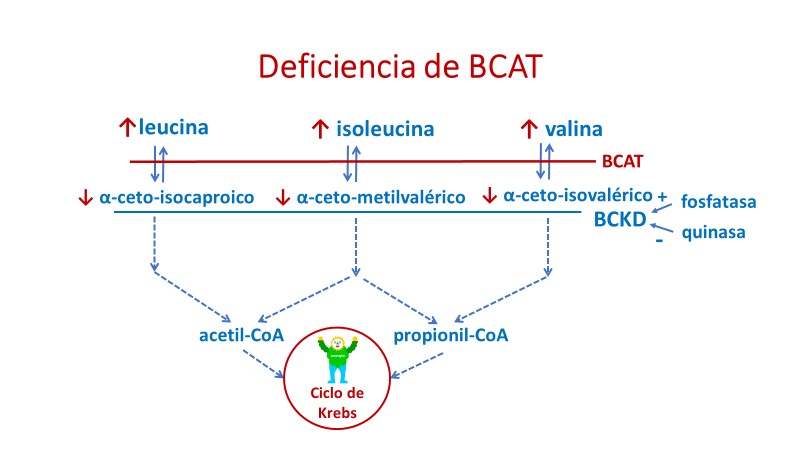
Deficiencia de la fosfatasa
La deficiencia de la fosfatasa responsable de la activación del complejo deshidrogenasa de los cetoácidos ramificados (BCKDK) es una nueva enfermedad metabólica diagnosticada por cribado neonatal en una paciente con moderada elevación de los AAR, aloisoleucina y cetoácidos ramificados (Oyarzábal y col, 2012, Centro de Biología Molecular Severo Ochoa CSIC-UAM, Universidad Autónoma de Madrid). La paciente ha sido tratada con dieta baja en proteínas, tiene actualmente 21 años y es enfermera.
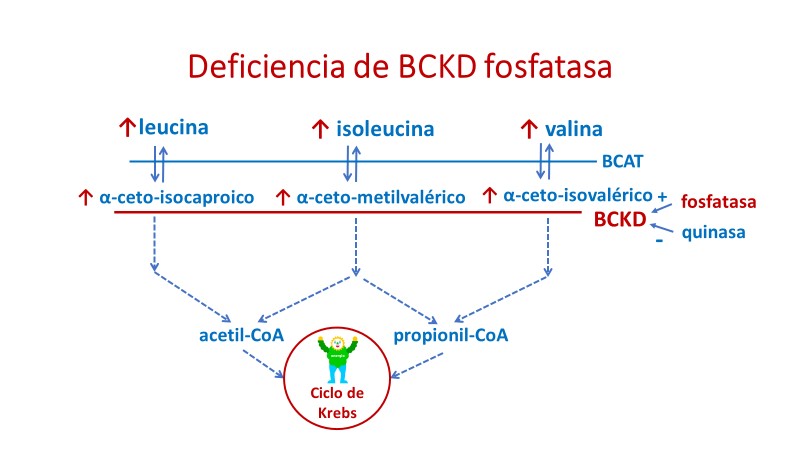
Deficiencia de la quinasa
La deficiencia de la quinasa responsable de la inactivación del complejo deshidrogenasa de los cetoácidos ramificados (BCKDK) es una nueva enfermedad metabólica de reciente descripción (Novarino y col, 2012, University of California, San Diego, USA; García-Cazorla y col, 2014, Hospital Sant Joan de Déu, Barcelona).
Mutaciones en el gen BCKDK interfieren en la inactivación del complejo BCKD causando su activación, lo que da lugar a una gran disminución de los niveles de los AAR y cetoácidos ramificados en sangre, orina y LCR al aumentar su catabolismo.
Los primeros dos pacientes descritos mostraban hipotonía, retraso del crecimiento, microcefalia y trastornos de conducta.
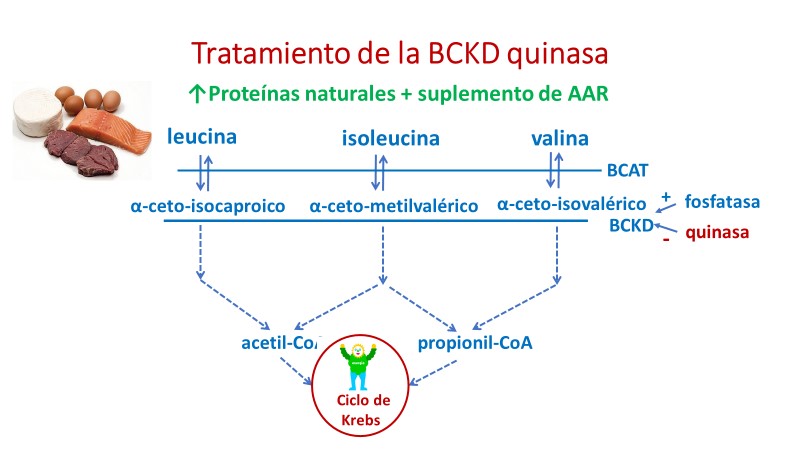
El tratamiento con una dieta rica en proteínas y suplemento de AAR normalizó los niveles plasmáticos de los mismos y mejoró el crecimiento, las variables de desarrollo y comportamiento.
Se han diagnosticado hasta el momento en Europa 20 pacientes y se ha iniciado un estudio colaborativo dentro del Proyecto MetabERN (Drs. T. Tangeraas y A. García-Cazorla) para conocer la historia natural de esta enfermedad ya que es un defecto metabólico tratable.
Conclusiones generales
La vía común de degradación de los AAR se ha podido estudiar con mayor detalle debido a los nuevos defectos metabólicos descritos recientemente en ella.
Estos defectos señalan la importancia no solo de la acumulación de metabolitos potencialmente tóxicos, en que se basan la mayoría de las enfermedades estudiadas, sino también el defecto de los productos de una reacción inhibida, que puede causar daños cerebrales más sutiles, pero igualmente importantes.
La reciente descripción de estos defectos menos graves y potencialmente tratables va a permitir conocer mejor la relación entre la alteración metabólica y sus consecuencias a nivel cerebral, que dan lugar a síntomas neurológicos o neuropsicológicos.
La extensión del cribado neonatal en la mayor parte de países permite diagnosticar prematuramente los defectos clásicos como la enfermedad de jarabe de arce, mejorando su pronóstico.
Además, permite descubrir defectos nuevos cuyas alteraciones bioquímicas son más sutiles y cuya clínica es menos grave, sin descompensaciones clínicas que sugieran una enfermedad metabólica hereditaria y aconsejen su investigación.
Muchas de estas enfermedades son potencialmente tratables y los pocos casos descritos sugieren una mejora clínica y una normalización bioquímica, lo que aumenta el interés del diagnóstico y seguimiento de estos pacientes.
- 1021 lecturas
También te puede interesar
: enfermedad de jarabe de arce")

