Diagnóstico bioquímico/genético

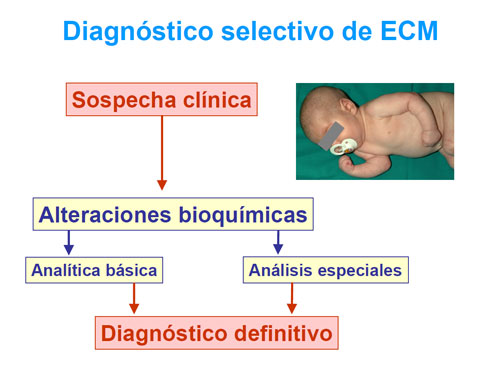
El diagnóstico de un ECM puede ser precoz neonatal masivo a toda la población o selectivo, basado en la sospecha clínica.
El diagnóstico precoz masivo (cribado neonatal o detección precoz neonatal) se realiza en la mayor parte de países del mundo siguiendo unos criterios establecidos (WHO, 1968; National Academy of Science, 1970).
Descrito por Guthrie en 1965 para la fenilcetonuria (PKU), incluye desde 2 (cribado neonatal convencional para fenilcetonuria e hipotiroidismo) hasta unas 30 enfermedades metabólicas (cribado neonatal extendido, de reciente implantación en algunos países o Comunidades Autónomas).
El diagnóstico selectivo se realiza analizando las alteraciones del metabolismo que muestran los pacientes, previamente seleccionados mediante la sospecha clínica.

Estas alteraciones se traducen generalmente en anomalías de los perfiles bioquímicos en líquidos biológicos (sangre, orina y, si es necesario, líquido cefalorraquídeo (LCR)), que orientan sobre las vías metabólicas afectadas y los puntos concretos de bloqueo, lo que permite confirmar los defectos mediante análisis enzimáticos o de caracterización de las proteínas mutadas y estudios genéticos.
Las muestras biológicas deben obtenerse, si es posible, en la descompensación metabólica (si la hay) y antes del tratamiento. Deben acompañarse de la información clínica sobre la edad, sexo, antecedentes familiares y personales de interés, breve resumen de la historia, dietas especiales y medicación.
Estos datos son indispensables porque el diagnóstico definitivo no deriva en general de un dato bioquímico aislado, sino de la interpretación del conjunto de datos bioquímicos y clínicos del paciente.
Por esto, la colaboración clínico-bioquímico es indispensable para la eficacia del diagnóstico de estas enfermedades.
Entre los análisis bioquímicos aplicados al diagnóstico de un ECM distinguimos unos procedimientos simples que constituyen el análisis básico de alteraciones metabólicas, que aporta una primera orientación sobre la vía afectada (glucosa, amonio, lactato).
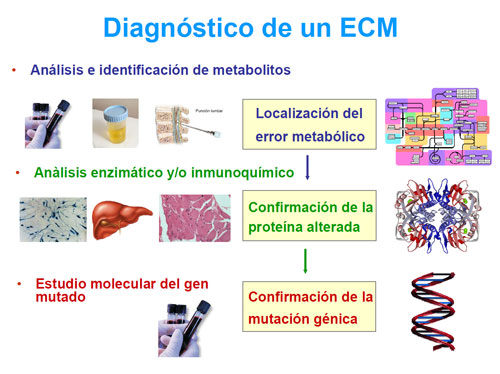
Ésta se confirma con la ayuda de otros análisis específicos, que se realizan, siempre que sea posible, en las mismas muestras de sangre y orina recogidas en descompensación y dependiendo de la sospecha clínica y de los datos aportados en los análisis básicos.
Si la combinación de los datos clínicos y bioquímicos ha permitido una orientación definitiva del defecto, se procede a la realización de estudios de confirmación de la proteína afectada por la mutación del DNA que constituye el origen de la enfermedad.
Estos estudios de confirmación pueden realizarse en suero, leucocitos, eritrocitos, fibroblastos cultivados procedentes de biopsia de piel o en tejidos (músculo, hígado, riñón, corazón, etc…). Los estudios genéticos de las mutaciones implicadas en el defecto completarán el estudio. Para ellos se extrae DNA de sangre periférica o tejidos del paciente.