Clasificación Internacional de las Enfermedades Metabólicas Hereditarias (ICIMD)
")
Durante la última década se han realizado diversas iniciativas de clasificación, como las basadas en un enfoque patofisiológico, de gran utilidad en la práctica clínica (Saudubray y col, 2019). Pero el enorme crecimiento del número de trastornos metabólicos hereditarios descritos en los últimos años (más de 1.500 según la base de datos IEMbase, debido a las nuevas tecnologías (bioquímicas y genéticas) que han permitido su diagnóstico, ha determinado la utilidad de una clasificación internacional basada en las vías metabólicas afectadas.
Además, este crecimiento ha conllevado la necesidad de definir un nuevo concepto de trastorno metabólico hereditario (Morava y col, 2015). Esta definición amplia mucho el número de trastornos metabólicos y determina la necesidad de una nueva clasificación que incluya todos los descritos y que permita la inclusión de los nuevos, que previsiblemente se describirán en un futuro más o menos próximo.

¿Cuál es el objeto de una clasificación internacional?
La clasificación internacional de las enfermedades metabólicas hereditarias (EMH) tiene por objeto combinar el conocimiento y la opinión de un gran número de especialistas e investigadores de todo el mundo en dichos trastornos para realizar una clasificación, que mantenga una estructura estable a largo plazo y que tenga el respaldo de las sociedades profesionales internacionales.
Además de proporcionar una lista completa de enfermedades, su objetivo es facilitar una mejor comprensión de las interconexiones entre las condiciones que comparten características funcionales, clínicas y de diagnóstico.
¿Cuál es la estructura general de esta clasificación?
Las EMH están divididas en 24 categorías que comprenden 124 grupos (ver Tabla 1).
| Tabla 1. Clasificación international de Enfermedades Metabólicas Hereditarias | ||
| Metabolismo intermediario: nutrientes |
1.Aminoácidos |
orgánicas, aa. ramificados, aa. aromáticos, aa. sulfurados, Gly y Ser, Orn, Pro y Hpro, Lys, HLys y Tpr, Glu/Gln y Asp/Asn y transporte aminoácidos. |
|
2.Péptidos y aminas |
glutationa, poliaminas y otros. | |
| galactosa y fructosa, Gluconeogénesis, Glucolisis, Glucógeno, pentosas y polioles, transporte y absorción de carbohidratos. | ||
| carnitina, oxidación mitocondrial de ácidos grasos, cuerpos cetónicos. | ||
| Metabolismo intermediario: energía | 5.Sustratos energéticos |
piruvato, ciclo de Krebs, creatina. |
| 6.mtDNA | proteínas de la cadena respiratoria, tRNA/rRNA. | |
| 7.Fosforilación oxidativa de origen nuclear |
subunidades de los Complejos I, II, III, IV y V, y factores de ensamblaje. |
|
| 8.Biosíntesis de cofactores | Coenzima Q10, ácido lipoico y hierro/azufre. | |
| 9.Mantenimiento y replicación del DNA |
pool de nucleótidos, mtDNA. |
|
| 10.Expresión genética mitocondrial | mt- aminoacil-tRNA sintetasas, mitoribosomas, etc. | |
| 11. Otros trastornos de la función mitocondrial: transporte... | ||
|
12.Trastornos reparación de metabolitos |
||
| 13.Miscelánea de trastornos del metabolismo intermediario: oxalato, glioxilato | ||
| Metabolismo y transporte de lípidos | 14.Lípidos |
síntesis, elongación y reciclaje de ácidos grasos (AG), oxidación peroxisomal de AG, eicosanoides, glicerolípidos. Glicerofosfolípidos. |
| 15.Lipoproteínas | síntesis y reciclaje de esfingolípidos, esteroles, ácidos biliares, hipercolesterolemias, hipertrigliceridemias, hiperlipidemias, metabolismo del HDL, LDL/triglicéridos, etc. | |
| Metabolismo de compuestos heterocíclicos |
16.Nucleobases, nucleótidos y ácidos nucleicos |
pirimidines, purines, ectonucleótidos y ácidos nucléicos, tRNA no mitocondrial, biogènesis de ribosomes. |
| 17.Tetrapirroles | síntesis de Hemo y porfirias, degradación Hemo y bilirubina. | |
| Moléculas complejas y metabolismo de orgánulos | 18.Defectos congénitos de la glicosilación (CDG): | N-glicosilación de proteínas, O-glicosilación de proteínas, glicosilación de lípidos, múltiples vías de glicosilación (dolicol, Golgi y otros). |
| 19.Biogénesis, dinàmica e interacciones de orgánulos | biogènesis peroxisomal, trafico vesicular y otros. | |
| 20.Degradación de moléculas complejas | autofagia, lipofuccinosis ceroidea, degradación de lipoproteínas, glucosaminoglicanos y esfingolípidos. | |
| Metabolismo de cofactores y minerales |
21.Elementos traza y metales |
hierro, cobre, cinc, manganeso, otros. |
| 22.Vitaminas y cofactores |
colalamina, folato, piridoxina, biotina, pantotenato y CoA, niacina y NAD, riboflavina, tiamina, tetrahidrobiopterina, cofactor molibdeno.
|
|
| Señalizadores metabólicos celulares |
23.Metabolismo de neurotransmisores |
ciclo de vesículas sinápticas, colina, glicina, glutamato, GABA, monoaminas. |
|
24.Metabolismo endocrino |
esteroides, insulina. | |
¿Cómo se relacionan entre sí estas categorías y subcategorías?
Tienen una funcionalidad interactiva mediante un diagrama que permite una visualización intuitiva de los diferentes grupos y subgrupos. Al hacer clic en ellos, se abre la tabla de enfermedades respectiva, con hipervínculos a otras bases de datos, y unas tablas que incluyen todas las enfermedades metabólicas hereditarias.
Diagrama de clasificación internacional de EIM
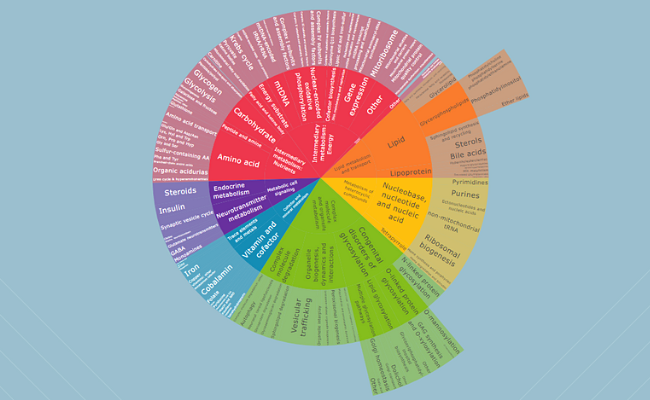
Clasificación basada en la fisiopatología de las EMH (Saudubray y col, 2019)
Esta clasificación, que se ha ido ampliando a medida que se han descrito nuevas enfermedades, es muy útil en la práctica clínica, porque incluye fenotipos clínicos y mecanismos de enfermedad agrupados en tres grandes categorías, por lo que se considera simplificada.
Como la mayoría de los tratamientos se basan en la fisiopatología de estas enfermedades, esta clasificación tiene la ventaja que vincula síntomas, fisiopatología y manejo terapéutico. Se basa en el tamaño de las moléculas (pequeñas o complejas) y su implicación en el metabolismo energético.
Los trastornos de moléculas pequeñas se resumen en la Tabla 2.
La acumulación de moléculas pequeñas causa trastornos por “intoxicación” aguda, intermitente, crónica o incluso progresiva. Los signos y síntomas resultan de la acumulación anormal de los compuestos proximales al bloqueo. No interfieren en el desarrollo embrionario y fetal y se presentan tras un intervalo sin síntomas con signos de intoxicación, provocada por el ayuno, el catabolismo, fiebre, enfermedades intercurrentes y excesiva ingesta de alimentos. La mayoría de estos trastornos son tratables y requieren la eliminación de la “toxina” mediante dietas especiales, depuradores y cofactores (vitaminas).
Los trastornos debidos a la deficiencia de moléculas pequeñas se deben a la síntesis defectuosa de compuestos esenciales distales al bloqueo o al defecto de transporte de moléculas esenciales a través de las membranas celulares u organelas. La mayoría afectan al desarrollo neurológico y pueden presentarse prenatalmente. Algunos de ellos son tratables suministrando el compuesto deficiente.
|
Tabla 2. EMH de las pequeñas moléculas |
||
| Características principales | Acumulación: intoxicación | Déficit |
| Acumulación de metabolitos próximos al bloqueo enzimático | Déficit de síntesis o transporte de una molécula esencial | |
| Sin repercusión en el desarrollo embriofetal | ||
| Intervalo libre de síntomas (de días a años en formas tardías | Interfiere en el desarrollo embriofetal (puede presentar malformaciones al nacimiento) | |
| Signos clínicos de intoxicación (episodios agudos, intermitentes, crónicos, o progresivos), desencadenados por fiebre, infecciones, ayuno prolongado o ingesta excesiva de proteínas | Los mecanismos de enfermedad en moléculas esenciales y no esenciales son diferentes | |
|
EMH implicadas |
EMH de aminoácidos (PKU, MSUD), defectos del ciclo de la urea, acidurias orgánicas, intoxicaciones por carbohidratos (fructosa, galactosa), porfirias, metales, purinas y pirimidinas (algunos) |
EMH de síntesis o transporte de aminoácidos (serina, glutamina, asparagina) Déficit de transportadores EIM que causan déficit de metales (Zn, Cu, Mn) EIM de neurotransmisores Defectos de vitaminas, purinas y pirimidinas (algunos) |
| Biomarcadores |
La mayoría tienen biomarcadores que permiten el diagnóstico y la monitorización del tratamiento |
|
| Manifestaciones neurológicas | Intoxicación neurológica aguda: encefalitis, ataxia, trastornos del movimiento, convulsiones | Encefalopatías precoces: microcefalia, retraso psicomotor, disfunción motora, etc... |
| Signos crónicos: dificultades de aprendizaje, síntomas psiquiátricos, paraparesia espástica | ||
|
Neurodegeneración: encefalopatías progresivas |
||
| Tratamiento | La mayoría tratables (dietas especiales, depuradores, cofactores) | Tratables, administrando el compuesto que falta |
Los trastornos de moléculas complejas (Tabla 3) alteran el metabolismo de dichas moléculas. Tienen lugar en todas las organelas (mitocondrias, lisosomas, peroxisomas, retículo endoplasmático, aparato de Golgi, gotas lipídicas, vesículas sinápticas). La mayoría no tienen un biomarcador bien identificado, con lo que su diagnóstico se basa en técnicas moleculares. En general, son enfermedades neurológicas progresivas que suelen asociar manifestaciones sistémicas.
Los defectos catabólicos conducen al almacenamiento de compuestos anormales que dan como resultado los defectos lisosomales clásicos (es decir, esfingolipidosis, muchas polisacaridosis).
Los trastornos de síntesis, remodelación, tráfico, procesamiento y control de calidad de moléculas complejas se han incluido más recientemente en este grupo. Estas enfermedades tienen un gran impacto tanto en la neurología infantil como en la del adulto. Se encuentran en la encrucijada entre los IEM tradicionales y las enfermedades genéticas, incluyen trastornos clásicos de orgánulos y diversos mecanismos de biología celular.
|
Tabla 3. EMH de las moléculas complejas: características fisiopatológicas |
|
| 1.Defectos del catabolismo | Causan el acúmulo de esfingolípidos o mucopolisacáridos: enfermedades lisosomales clásicas: mucopolisacaridosis, esfingolipidosis... |
| 2.Defectos de síntesis, remodelación, procesamiento, tráfico y control de calidad |
Defectos clásicos: Enfermedades lisosomales y peroxisomales |
|
Defectos de la glicosilación de las proteínas (CDG) |
|
|
Defectos de síntesis de colesterol y ácidos biliares |
|
|
Defectos de síntesis de glicosaminoglicanos |
|
|
Defectos de síntesis y remodelación de lípidos complejos (triglicéridos, fosfolípidos,...) |
|
|
Defectos de síntesis y transporte de ácidos grasos |
|
|
Defectos de procesamiento, tráfico y control de calidad (vesiculación intracelular, tráfico, reparación, vesícula sináptica) |
|
Los defectos relacionados con la energía son debidos, al menos en parte, a un déficit en la producción de energía o a la utilización de la misma en cerebro, músculo, miocardio, hígado y otros tejidos (Tabla 4).
El diagnóstico puede orientarse mediante pruebas funcionales que miden glucosa, lactato, cetonas y otras moléculas energéticas en sangre, orina y líquido cefalorraquídeo, debiendo confirmarse mediante análisis enzimáticos y genéticos.
Las principales presentaciones neurológicas de estas enfermedades son:
- Encefalopatías graves neonatales con acidosis láctica, a menudo asociadas a síntomas multisistémicos.
- Síntomas neurológicos agudos en momentos de alta demanda energética.
- Signos motores crónicos: trastornos del movimiento, ataxia, neuropatía periférica.
- Epilepsia mioclónica, ausencias atípicas.
- Afectación neurosensorial.
| Características fisiopatológicas | |
| 1.Transportadores de moléculas energéticas | Glucosa, ácidos grasos, cuerpos cetónicos, ácidos monocarboxílicos, presentan isoenzimas específicas, como los defectos de GLUT1 y GLUT2. |
| 2.Defectos mitocondriales | Oxidación de los ácidos grasos y de cuerpos cetónicos. Defectos de la carnitina. |
| Déficits de la oxidación aeróbica de la glucosa, que se presentan como acidosis lácticas (defectos del piruvato y ciclo de Krebs). | |
| Defectos de la cadena respiratoria mitocondrial. | |
|
Defectos de síntesis de coenzima Q10. |
|
| Transportadores mitocondriales de moléculas energéticas. | |
|
Defectos de la maquinaria mitocondrial (fusión, fisión, replicación, importación de proteínas mitocondriales, control de calidad, ribosomopatías, depleción de ADN mitocondrial, etc. |
|
| 3.Defectos energéticos citoplasmáticos | Defectos de la glicolisis, metabolismo del glucógeno y glucogénesis, hiperinsulinismos, transportadores de glucosa, metabolismo de la creatina, etc. |