Mutaciones en CAD y encefalopatía epiléptica sensible a la uridina
Resumen de la Cápsula Metabólica presentada por la doctora Alejandra Darling en la Unidad de Enfermedades Metabólicas del Hospital Sant Joan de Déu de Barcelona, el 19 de junio de 2020.

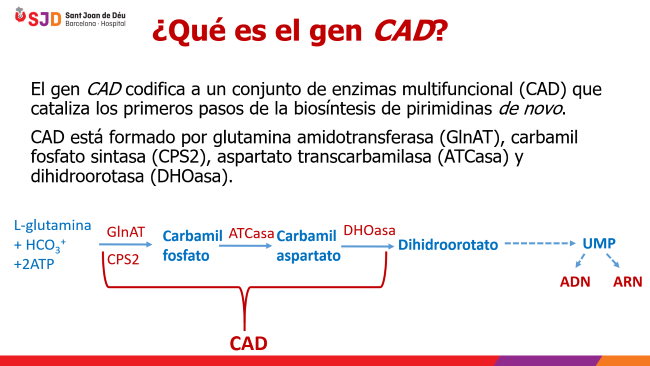
El gen CAD codifica un complejo enzimático multifuncional que cataliza los primeros pasos de la biosíntesis de pirimidinas de novo (es decir, mediante un proceso por el cual unas moléculas precursoras simples se convierten en productos más complejos a través de múltiples pasos catalizados por enzimas).
¿Qué son las pirimidinas?
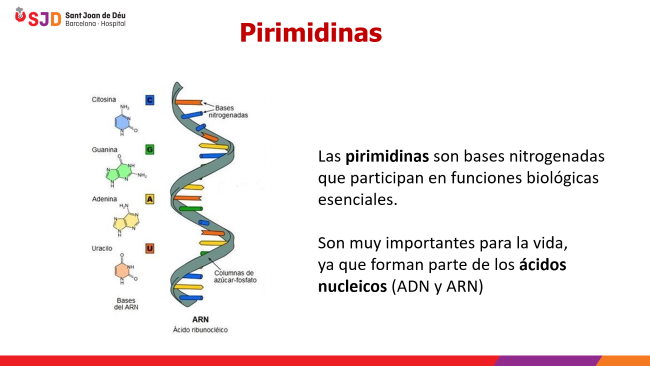
Las pirimidinas son bases nitrogenadas que participan en funciones biológicas esenciales. Tres de ellas son muy importantes para la vida, ya que forman parte de los ácidos nucleicos (ADN y ARN): la timina, la citosina y el uracilo; las dos primeras forman parte del ADN donde se aparean con sus purinas complementarias, mientras que la última está presente solo en el ARN.
¿Cuál es la función del complejo CAD?
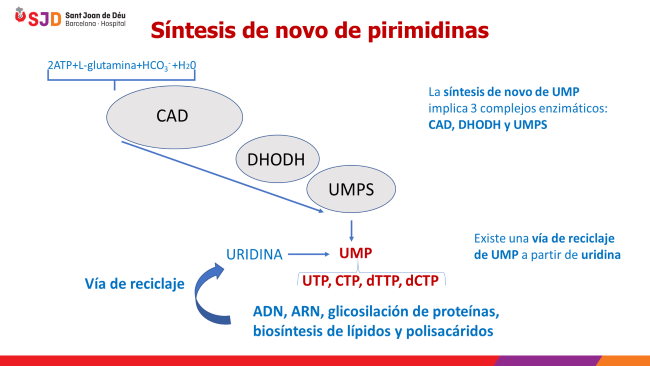
En la imagen superior vemos como a partir de moléculas sencillas (L-glutamina, bicarbonato y agua), con la aportación de energía (2ATP) y mediante la acción de CAD, otra enzima (DHODH) y otro complejo enzimático (UMPS) se forma uridina monofosfato (UMP), precursora de los nucleótidos (uridina trifosfato, citosina trifosfato, desoxiuridina trifosfato y desoxicitosina trifosfato) pilares básicos del ARN y ADN).
Existe una vía de reciclaje de UMP a partir de la degradación del ARN.
¿Qué ocurre si el complejo CAD no funciona correctamente?
Si el complejo CAD no funciona correctamente debido a mutaciones en el gen CAD, la síntesis de UMP no se realiza con la debida eficacia y todas las funciones biológicas que derivan de ella se ven comprometidas.
Dada la enorme importancia de dichas funciones, esto tiene consecuencias graves en el paciente que porta mutaciones en CAD.
¿Qué manifestaciones clínicas presentan los pacientes con deficiencia de CAD?
Los pacientes con deficiencia de CAD presentan un trastorno neurometabólico que incluye retraso global del desarrollo, convulsiones y anemia diseritropoyética (afecta el desarrollo de los glóbulos rojos de la sangre (eritropoyesis). El curso natural de la enfermedad puede ser letal ya desde la primera infancia.

¿Cuántos pacientes se han diagnosticado con deficiencia de CAD?
Se han diagnosticado hasta el momento 5 pacientes con deficiencia de CAD. Un primer paciente (descrito en 2015 por Ng y col) y 4 pacientes pertenecientes a tres familias (descritas por Koch y col en 2017).
La presentación clínica de los pacientes es similar y consiste básicamente en retraso global del desarrollo, convulsiones y anemia diseritropoyética (que afecta el desarrollo de los glóbulos rojos de la sangre (es decir, la eritropoyesis).
¿Cómo se realiza el diagnóstico de los pacientes?

Todos los análisis especiales para descartar una enfermedad metabólica fueron normales, a excepción de la electroforesis en gel de poliacrilamida de las proteínas de glóbulos rojos, que migraron anormalmente lo que sugirió una glicosilación anormal (según NG y col).
Los análisis bioquímicos específicos no identificaron un trastorno de pirimidinas, probablemente porque los metabolitos de pirimidina en la orina muestran una amplia gama de valores normales, lo que refleja las contribuciones de la dieta y de la degradación celular.

¿Tiene tratamiento esta enfermedad?
El hecho de que la síntesis de uridina monofosfato (UMP) tenga una vía alternativa de reciclaje sugirió a Koch y sus colaboradores que el tratamiento con uridina podía ser efectivo.
Dos pacientes fueron tratados a la edad de 5 y 7 meses con uridina oral y ambos respondieron con una notable mejoría clínica.
Esto apoya el potencial de la administración de uridina para mejorar una enfermedad neurodegenerativa severa que puede ser letal en la infancia.
Factores que considerar en el diagnóstico de esta enfermedad
- La anemia es normocítica (es decir, que el volumen corpuscular medio es normal). No obstante, se observa una distribución anormal del tamaño y morfología de los eritrocitos: por lo que se recomienda realizar un frotis y comprobar la curva de distribución del tamaño en el contaje.
- A pesar de que la deficiencia de CAD es un trastorno primario de la biosíntesis de pirimidinas de novo con la glicosilación secundariamente alterada, las pruebas metabólicas específicas para estos trastornos (es decir, medición de pirimidinas y el isoelectroenfoque de transferrina) son normales.
- La atrofia cerebral progresiva que es una característica consistente en la neuroimagen es bastante inespecífica y solo se puede observar en una etapa tardía de la enfermedad; las resonancias magnéticas tempranas eran normales.
- Se debe tener en cuenta que los factores nutricionales pueden modificar la gravedad y el curso de la deficiencia de CAD y podría enmascarar datos fenotípicos indicativos (por ejemplo, eritrocitos).

Referencias:
Ng BG, Wolfe LA, Ichikawa M, Markello T, He M, Tifft CJ, Gahl WA and Freeze HH. Biallelic mutations in CAD, impair de novo pyrimidine biosynthesis and decrease glycosylation precursors. Hum Mol Genet 2015;24(11):3050-7.
Koch J, Mayr JA, Alhaddad B y col. CAD mutations and uridine-responsive epileptic encephalopathy. Brain Advance Access published December 21, 2016: 1-8.
Por otra parte, en Guía metabólica pueden hallar más información: Defectos del metabolismo de las purinas y pirimidines.
- 1295 lecturas
También te puede interesar


