Deficiencia de acil-CoA deshidrogenasa de cadena muy larga (VLCAD)

La VLCAD es un error congénito del metabolismo de los ácidos grasos de cadena larga (de 14 a 20 átomos de carbono). Está causado por la deficiencia de la enzima acil-CoA deshidrogenasa de cadena muy larga (VLCAD), que cataliza el primer paso de la β-oxidación de los ácidos grasos.
Causa un bloqueo en la oxidación de ácidos grasos de cadena larga (de 14-20 átomos de carbono). La VLCAD controla un punto crítico en el suministro de electrones a la cadena respiratoria y además, controla la ruta de formación de cuerpos cetónicos.
Esto hace que, en condiciones de descompensación metabólica, se acumulen dichos ácidos, así como sus derivados conjugados con la carnitina (acilcarnitinas) y ácidos dicarboxílicos en sangre y orina.
¿Cuáles son las manifestaciones clínicas de la deficiencia de VLCAD?
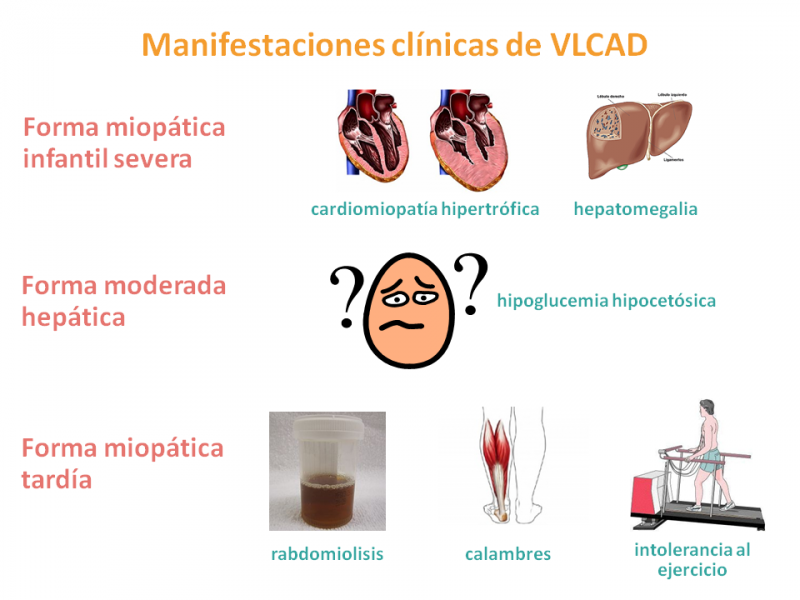
Se han descrito tres fenotipos (conjunto de características físicas, bioquímicas, fisiológicas, signos y síntomas de un individuo) diferentes:
- Forma infantil severa miopática con fallo multiorgánico, que se presenta en los primeros meses de vida con cardiomiopatía (enfermedad del corazón) hipertrófica (por engrosamiento) o dilatada (por debilidad), derrame pericárdico (acumulación de líquido en la bolsa serosa que rodea al corazón) y arritmias (alteraciones del ritmo cardíaco), así como hipotonía, hepatomegalia (hígado grande) e hipoglucemia intermitente.
La primera descompensación metabólica, generalmente antes de los 8 meses de edad, puede tener un final fatal. No obstante, la disfunción cardíaca es reversible con un diagnóstico y tratamiento intensivo precoz y modificación de la dieta. - Forma moderada hepática con hipoglucemias hipocetósicas, de presentación más tardía, en la infancia, con hepatomegalia y sin cardiomiopatía.
- Forma miopática tardía, del adolescente o adulto, se presenta con rabdomiolisis (lesión de los músculos esqueléticos) intermitente, con calambres musculares y/o dolor e intolerancia al ejercicio. Es progresiva e inducida por el ejercicio, ayuno o estrés, sin afectación cardíaca ni hipoglucemia.
- Muchos de los niños diagnosticados por cribado neonatal y tratados adecuadamente se han mantenido asintomáticos durante años, y este hecho, aunque muy importante, no permite asegurar que el tratamiento preventivo va a impedir la aparición de síntomas de por vida.
¿Cómo se diagnostica la deficiencia de VLCAD?
El diagnóstico se realiza en base a la presentación clínica o mediante el cribado neonatal ampliado a los defectos de la β-oxidación de los ácidos grasos.
El estudio de ácidos orgánicos en orina muestra un perfil característico de ácidos dicarboxílicos y 3-hidroxidicarboxílicos durante los períodos de descompensación metabólica.
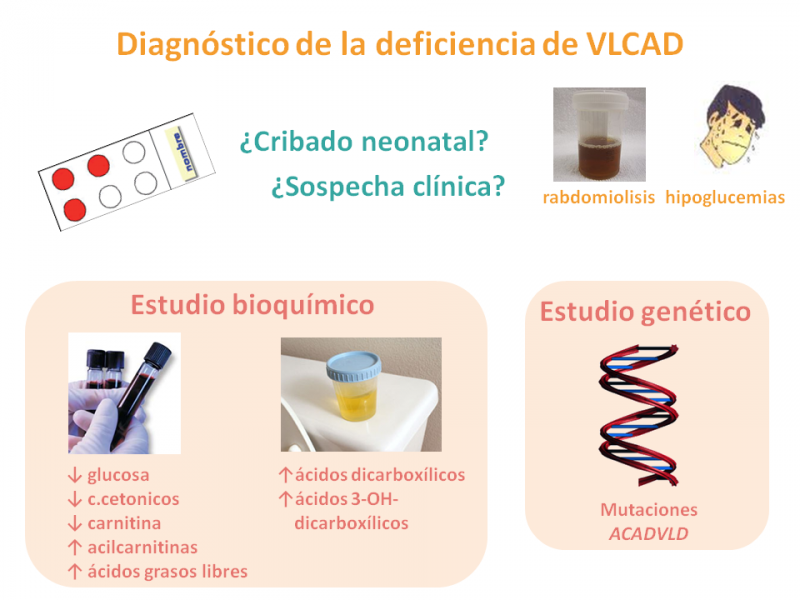
En sangre, los ácidos grasos libres y las acilcarnitinas específicas (C14) están elevados, mientras que se observa una acidosis láctica y una deficiencia de carnitina secundarias, incremento variable de enzimas hepáticas y elevación de CPK.
La hipoglucemia hipocetósica se observa en la forma hepática.
El cribado neonatal para la deficiencia de VLCAD, con inicio de un tratamiento adecuado, previene muchas de las descompensaciones y sus posibles secuelas, por lo que se está aplicando ya actualmente en muchos países.
El diagnóstico se confirma mediante el estudio de la oxidación de palmitato marcado en cultivo de fibroblastos y especialmente, mediante el estudio genético.
El espectro de mutaciones en el gen ACADVLD es heterogéneo, habiéndose descrito más de 60 mutaciones, ninguna prevalente.
El estudio genético permite el consejo genético familiar y el diagnóstico prenatal, si se requiere.
¿Tiene tratamiento la deficiencia de VLCAD?
El tratamiento común a todos los defectos de la β-oxidación se basa en prevenir la hipoglucemia, lo que se consigue:
- Evitando el ayuno prolongado, mediante una dieta fraccionada.
- Utilizando una dieta rica en hidratos de carbono, usando hidratos de carbono de absorción lenta (ver Consejos para evitar la hipoglucemia).
- Tratamiento dietético específico para VLCAD:
- Suprimir la leche materna y sustituirla por una fórmula especial suplementada en MCT
- Restringir LCT (triglicéridos de cadena larga).
- Suplementar con aceite de soja como fuente de precursores de ácidos grasos esenciales, para evitar su defecto.
- Suplementar con MCT previa al ejercicio (en pacientes con posible rabdomiolisis).

Ante situaciones de stress (infecciones, cuadros febriles) evitar ayuno prolongado asegurando una ingesta adecuada de hidratos de carbono (a base de bebidas o alimentos ricos en hidratos de carbono).
Aconsejamos consultar la Pauta de descompensación en un defecto de la β-oxidación y metabolismo de la carnitina.
Consejos

Actualidad

Nutrición

Recursos

Todas las personas somos distintas, por dentro y por fuera. Sí, unas son morenas y tienen las piernas largas; otras son rubias y redonditas; algunas tiene orejas pequeñas y otras, los ojos como platos.
Eso es lo que podemos ver exteriormente. Pero en el interior de nuestro...

Una nueva aventura de la Pandilla Metabólica, en la que Annia, Tomás, Laura, Gabriel y Arnau tienen dos misterios para resolver.
Desde hace un tiempo, la clase de al lado saca mucho mejores notas que ellos en los exámenes. Su profesora no está nada contenta y les echa...

La Pandilla Metabólica va haciendose mayor y ya han acabado sus estudios de secundaria. Laura y Ámbar han empezado sus estudios en la Escuela de Hostelería, su gran ilusión desde pequeñas.
Pero al llegar se encuentran con que no hay ninguna asignatura especializada...

La aplicación Primeros Auxilios Fáciles proporciona, de una manera clara y sencilla, conocimientos básicos y pautas concretas para aplicar medidas de primeros auxilios en caso de urgencia antes de...

Con esta aplicación podrás pintar infinidad de mandalas y trabajar la motricidad fina y la coordinación ojo-mano, además de la creatividad y la imaginación.