Resumen de la XIX Jornada de pacientes y familias PKU-OTM del Hospital Sant Joan de Déu (2ª parte)
Resumen de la reunión online mantenida entre el Hospital Sant Joan de Déu (HSJD) y Hospital Clínic (HC), Barcelona, 17 de octubre de 2020.

La primera parte de la Jornada de pacientes y familias PKU-OTM, que trató temas de interés general, puedes encontrarla también en Guía metabólica.
Esta segunda parte de la jornada consistió en cuatro reuniones simultáneas para diferentes enfermedades metabólicas: fenilcetonuria, transportador de glucosa GLUT1, aciduria glutárica tipo 1 y enfermedades lisosomales.
Grupo de la fenilcetonuria (PKU)
El primero tema de esta reunión, presentado por la Dra. Sílvia Mediavilla (Gastro-Nutrición, HSJD) y la Sra. Natàlia Egea (Dietética-Nutrición, HSJD), consistió en una serie de preguntas por la app Kahoot para los adolescentes, adultos y padres de hijos afectos de PKU en relación con la alimentación, las proteínas de alto valor biológico y otros muchos aspectos nutricionales que deberían conocer.
Se trata de una serie de preguntas interactivas con 4 respuestas posibles, una de ellas verdadera, que los asistentes iban respondiendo y los ponentes analizando la respuesta correcta. Hubo una alta participación y después de 13 preguntas, resultó una vencedora, pero los demás no se quedaron atrás y brillaron también por sus conocimientos.

En la segunda parte el Dr. Campistol (Neuropediatría, HSJD) analizó aspectos novedosos en relación con la PKU como, por ejemplo, las últimas tendencias recogidas en los nuevos protocolos, que se publicaran en 2021 y en los que participa directamente la Unidad de Enfermedades Metabólicas Congénitas del Hospital Sant Joan de Déu Barcelona.
Se analizaron los últimos trabajos sobre la terapia enzimática con peg-valiasa, sus ventajas, los efectos secundarios y las posibilidades de empleo según edad y control de la enfermedad.
Están emergiendo terapias génicas para algunas enfermedades y la PKU no se queda atrás. La empresa Biomarin está iniciando un ensayo clínico para terapia génica en pacientes con PKU mayores de 18 años, y de confirmarse las buenas perspectivas en un futuro próximo se podrán aplicar a los menores de 18 años. Es un ensayo clínico y por tanto hay que esperar los primeros resultados, que de momento ya se han dado en ratones PKU.
Finalmente se comentó la aparición en Estados Unidos del genérico de Kuvan, que supondrá sin duda un importante ahorro para todos los pacientes en tratamiento, especialmente en los países donde no está cubierto por el sistema de salud.
Finalmente, en la tercera parte la Sra. Laia Molla (Psicología, HSJD) analizó los problemas del adolescente con PKU, en especial a nivel escolar y social. Como acepta la enfermedad, como se rebela ante la misma y todo lo que conlleva de análisis, visitas al hospital, dietas etc. Hubo un largo debate en este sentido y también con los niños más pequeños y los problemas que ya aparecen en la escuela. Se propuso usar el documento ya existente en Guía metabólica para informar a los maestros y compañeros. También se propuso grabar un vídeo para que los maestros y tutores puedan pasarlo en la clase donde haya un niño con PKU. Finalmente se acordó que en la próxima reunión se intentará elaborar un sistema de preguntas y respuestas a cargo de Psicología en relación con la PKU.
Transportador de glucosa (GLUT1)
En el grupo del transportador de glucosa (GLUT1), las Dras. Leticia Pías y Àngels García Cazorla (Neuropediatría, HSJD) hablaron de las epilepsias metabólicas, con especial atención al defecto de GLUT1.
La deficiencia de GLUT1 da lugar a un síndrome de insuficiencia energética cerebral causado por la alteración del transporte de glucosa a través de la barrera hematoencefálica y las membranas de los astrocitos. La dieta cetogénica es el tratamiento de elección en el defecto de GLUT1, ya que proporciona una fuente alternativa de energía (los cuerpos cetónicos) que elude la deficiencia de glucosa en el cerebro y mejora la mayor parte de los síntomas de deficiencia de GLUT1.
Los cuerpos cetónicos circulantes actúan como sustitutos de la glucosa al convertirse en acetil-CoA, que luego entra en el ciclo de Krebs y se oxida en las mitocondrias para producir energía (ATP).
La Dra. Pias comentó los estudios multidisciplinarios (Neuropediatría, Gastroenterología-Nutrición, Laboratorio de Metabolopatías) que se estaban llevando a cabo en grupos de pacientes con epilepsias metabólicas tratados con dieta cetogénica, así como otros proyectos de futuro.
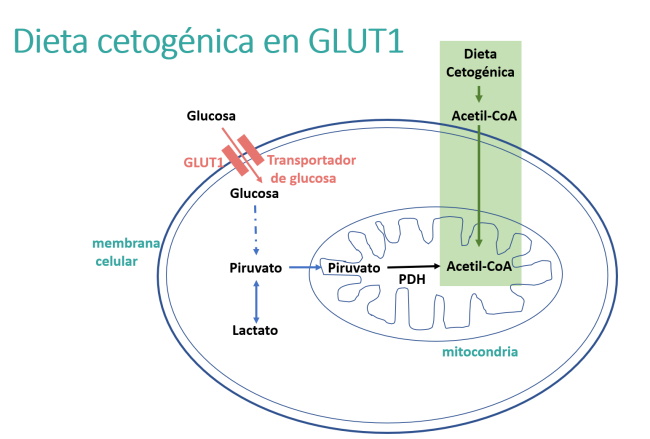
La dietista-nutricionista Lola García (HSJD), explicó en qué consiste el tratamiento con dieta cetogénica. Dicha dieta debe ser rica en grasa y reducida en carbohidratos, con la cantidad de proteína suficiente, que se calculará dependiendo del paciente de manera individual, con el fin de cambiar el sustrato energético de glucosa a cuerpos cetónicos. Precisamente estos cuerpos cetónicos, son los metabolitos beneficiosos responsables de la reducción de las crisis en epilepsia.
También describió los tipos de dietas cetogénicas que se indican en el Hospital Sant Joan de Déu, cómo se instauran y los controles a seguir, ya sea en el hospital o en el domicilio. Otro aspecto que expuso fueron las nuevas herramientas que existen para mejorar la adherencia a las pautas cetógenas como: la nueva masa madre de “pan cetógeno” de la Fundación Alicia, o las pastas a base de glucomanano.
Finalmente, hizo hincapié en que es un tratamiento, aunque sea una dieta, y es muy importante seguirlo de manera estricta, para obtener los beneficios esperados, por lo que necesita de un compromiso y una implicación firme por parte de las familias y cuidadores.
El Dr. Pere Leyes (Unidad de Adultos del Hospital Clínic) explicó la dieta cetogénica en el adulto con déficit de GLUT1. Como dicha dieta es un tratamiento a largo plazo, se deben vigilar y prevenir las posibles complicaciones.
La dieta cetogénica en la vida adulta comporta una serie de retos: el paciente adulto es/debe ser autónomo, por lo que debe hacer compatible la dieta cetogénica con su plan de vida. La transferencia del paciente adolescente al hospital de adultos implica unos cambios a un entorno menos protector y la fragmentación de la asistencia especializada.
Falta información de la evolución a largo plazo, que permita unas medidas preventivas adecuadas. Se debe hacer una adaptación a los requisitos del adulto y es importante mediterranizar la dieta. Se debe impulsar una transferencia de responsabilidad de los padres al paciente, implicando al mismo en su dieta.
Conviene hacer una revisión periódica de posibles complicaciones crónicas a medio y largo plazo: dislipemia (con riesgo cardiovascular), déficit de calcio y selenio (implica su suplementación), retraso del crecimiento (se debe ajustar el aporte proteico), litiasis renal y alteraciones óseas. Se debe realizar un control de la posible litiasis renal (análisis de orina y eventual tratamiento preventivo), de cardiopatía (ECG y eventual ecocardiografía) y alteraciones óseas (densitometría y estado de la vitamina D). Se debe considerar la suplementación (multivitamínica, calcio+ vitamina D), evaluación del riesgo cardiovascular (control de dislipemia y otros factores de riesgo) y del estado nutricional (carnitina, selenio).
Grupo de la Aciduria Glutárica tipo 1
Las Dras. Alejandra Darling (Neuropediatría, HSJD), Mariela de los Santos (Gastroenterología-Nutrición, HSJD) y la dietista-nutricionista Mireia Termes (HSJD) expusieron las novedades en el tratamiento de la Aciduria Glutárica.
Se realizó un resumen de distintos aspectos de la patología, haciendo hincapié en el tratamiento y las novedades en el tratamiento dietético. La Dra. Alejandra Darling hizo una introducción, explicando la causa de esta entidad, comenzando por el defecto genético y sus consecuencias a nivel bioquímico, celular y clínico. Se revisaron artículos recientes, que reflejan la importancia fundamental del cribado neonatal y las consecuencias en el control y la evolución de la enfermedad, que está cambiando favorablemente en los últimos años.
La Dra. Mariela de los Santos explicó las principales recomendaciones comentadas en la última guía revisada por el grupo multidisciplinar de expertos desarrollada en Heidelberg y publicada en el año 2016. La dietista-nutricionista Mireia Termes explicó el desarrollo de las nuevas pautas dietéticas que se están realizando en la Unidad de Enfermedades Metabólicas Congénitas, e ilustró las nuevas recomendaciones dietéticas utilizando ejemplos, además de comentar los cambios en la dieta según la edad.
Finalmente, se estableció un tiempo de diálogo con las familias, compartiendo su experiencia con la del equipo profesional.
Grupo de Enfermedades lisosomales
En el grupo de enfermedades lisosomales, la Dra. Mar O’Callaghan (Neuropediatría, HSJD) trató de las enfermedades lisosomales en edad pediátrica: nuevas terapias y traspaso a centro de adultos.
Señaló que, tanto en la edad pediátrica como en la adulta, estos pacientes requieren de un equipo multidisciplinar, pero, así como en la edad pediátrica la atención se centra en el diagnóstico, el acompañamiento y la búsqueda de tratamientos curativos, en el adulto se dirige más bien a la aceptación de la enfermedad y la cronicidad de la misma.
Explicó las distintas estrategias de tratamiento: sintomático, de soporte, trasplante de precursores hematopoyéticos (médula ósea, cordón umbilical), terapia enzimática sustitutiva endovenosa/intratecal, terapia de inhibidores de sustratos, tratamiento de estabilización enzimática – chaperonas y terapia génica.

El tratamiento sintomático sigue siendo el pilar en este grupo de enfermedades. La aparición progresiva de síntomas y la afectación multisistémica hacen necesaria la atención por parte de un equipo multidisciplinar, que conozca bien la historia natural de la enfermedad. Es importante el apoyo por parte de los equipos de crónicos y paliativos.
La única terapia potencialmente curativa es el trasplante de precursores hematopoyéticos (médula ósea o cordón umbilical). Es el tratamiento de elección en algunas enfermedades lisosomales (p.ej.: MPS II, MPS VII). Consigue recuperar la actividad de la enzima deficiente y en fases iniciales o presintomáticas de la enfermedad puede evitar el desarrollo de la misma.
La terapia enzimática sustitutiva es el tratamiento de elección para una gran parte de enfermedades lisosomales (MPS, CLN2, Gaucher). La terapia de reducción de sustratos se basa en el bloqueo de la síntesis de sustratos necesarios para la síntesis de la sustancia acumulada. El tratamiento de estabilización enzimática mediante chaperonas sintéticas que ayudan al correcto plegamiento de las enzimas ha sido aprobado en algunas enfermedades lisosomales (Fabry). Existe un número creciente de terapias génicas en fase de desarrollo y de ensayo en humanos. La Dra. O’Callaghan comentó la participación en ensayos clínicos en enfermedades lisosomales del HSJD, así como la derivación de pacientes del HSJD para tratamientos / ensayos en otros centros europeos.
El Dr. Milisenda (HC), hizo hincapié en el proceso de transición de los pacientes cuando llegan a la edad adulta. Ilustró en su presentación las instalaciones con las que cuenta el Hospital Clínic de Barcelona para atender los pacientes en diferentes ámbitos, como son consultas externas, hospital de día, sala de internación común y cuidados intensivos. También reflejó que el hospital cuenta con un gran equipo multidisciplinar, que interacciona en todo momento para mejorar la atención de los pacientes.

- 485 lecturas
También te puede interesar
")
")
