Epidemiología de las enfermedades metabólicas hereditarias (ECM)
")
La epidemiología es la disciplina que estudia los determinantes de la enfermedad y el impacto de los factores implicados en su desarrollo, distribución y diseminación en la población. Por lo tanto una parte muy importante de la epidemiología de una enfermedad es la que establece las causas de la misma, su desarrollo y su transmisión, es lo que se denomina epidemiología analítica.
Aunque posiblemente sea más conocida por el público la epidemiología descriptiva que es la que incluye los métodos para detectar y reconocer los casos, las medidas de frecuencia y su distribución en la población.
La aproximación a los ECM desde la perspectiva de su epidemiología tiene un impacto muy positivo para conocerlos, nos proporciona herramientas para detectar necesidades emergentes, mejorar la práctica asistencial y la toma de decisiones en política sanitaria.
Es decir, para poder planificar actividades preventivas y de control y promoción de la salud a fin de mejorar el pronóstico y la calidad de vida de los afectados, reducir la mortalidad y otras complicaciones devastadoras.
¿Cuál es la causa de los ECM, qué factores influyen en su desarrollo y cómo se transmiten? (Epidemiología analítica)
1. ¿Cuál es la causa de los ECM?
Los ECM están causados por variaciones llamadas mutaciones en alguno de los genes del ADN del núcleo celular. En unas pocas enfermedades específicas hay una contribución del ADN de la mitocondria.
Los genes codifican información para fabricar proteínas y las mutaciones en el ADN tienen como resultado un cambio en la proteína que puede afectar negativamente su función. Esto produce desequilibrios químicos en el organismo que inducen las manifestaciones clínicas.
2. ¿Qué factores influyen en el desarrollo de la enfermedad?
Los pacientes con una misma enfermedad metabólica hereditaria no son todos iguales y lo que aún es más intrigante es que en algunos casos, incluso hermanos con una misma mutación pueden mostrar distinta presentación y curso clínico.
En el desarrollo de la enfermedad influyen muchos factores internos y externos.
- Entre los internos están la heterogeneidad genética (en un gen puede haber mutaciones muy distintas que causan efectos de diferente importancia en la proteína) y también mutaciones en genes distintos pueden dar lugar a la misma enfermedad clínica.
Hay variaciones en el ADN en principio neutras, llamadas polimorfismos en vez de mutación, porque por si solas no producen enfermedad, pero situadas en el mismo gen oen otros genes pueden influir también en el cuadro clínico.

Otras causas de variabilidad clínica son variaciones en el número de copias de los genes, elementos móviles del genoma y factores relacionados con el “apagado” o “encendido” del gen y su expresión hasta llegar a la proteína (son los factores llamados epigenéticos porque están en realidad fuera del gen).
- Finalmente en el cuadro clínico pueden influir factores externos o medioambientales. Las mutaciones severas que alteran de manera importante la función de la proteína que es codificada por el gen, son causa suficiente de enfermedad, pero hay muchas mutaciones que permiten una función residual de la proteína.
En este caso, el curso clínico puede ser modificado por factores ambientales, como la fiebre, el estrés metabólico, medicamentos, entre otros. Por ejemplo, la ingestión de determinados medicamentos o de habas induce importantes crisis hemolíticas en personas con deficiencia de glucosa-6-fosfato deshidrogenasa. El ayuno prolongado en niños con trastornos de la beta-oxidación puede ser fatal.
3. ¿Cómo se transmiten las enfermedades metabólicas hereditarias?
Dado que están causadas por mutaciones en un único gen, se transmiten a la descendencia con un patrón de herencia llamado monogénico o mendeliano, porque sigue las llamadas leyes de Mendel.
La gran mayoría se transmiten con un patrón llamado recesivo, porque solo cuando ambos progenitores llevan una mutación en el mismo gen y las transmiten conjuntamente tendrán un hijo afectado.
Siguen un patrón llamado dominante cuando es suficiente que solo uno de los progenitores transmita la mutación para que el hijo lo esté.
El llamado patrón de herencia ligado al sexo es el que siguen las mutaciones en el cromosoma X, únicamente pueden padecer la enfermedad los hijos varones que reciben la mutación en su único cromosoma X, pero las niñas, que tienen dos cromosomas X, mantienen funcional uno de ellos y solo en ocasiones presentan síntomas clínicos.
Finalmente existe el llamado patrón de herencia materno o mitocondrial, que es el que siguen las mutaciones en el pequeño genoma mitocondrial, porque este genoma lo heredamos siempre de la madre.
¿Cómo se detectan los casos de una enfermedad, cómo se estima su frecuencia en una población y qué medidas de prevención y control se pueden adoptar? (Epidemiología descriptiva)
1. ¿Cómo se pueden discernir o detectar los casos de enfermedad metabólica hereditaria?
Generalmente a través del diagnóstico de los pacientes sintomáticos, mediante la sospecha clínica y su diagnóstico diferencial y empleando tecnologías de soporte, como los estudios por la imagen (radiografías, resonancia nuclear magnética y otras) y las investigaciones anatomopatológicas.
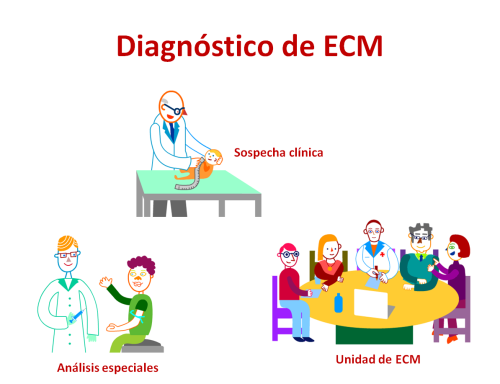
Pero dado que son enfermedades genéticas, las pruebas de laboratorio son el estándar de oro para un diagnóstico inequívoco y definitivo (son las pruebas de genética bioquímica y genética molecular).
Las pruebas de laboratorio serán también imprescindibles para el diagnóstico de heterocigotos portadores, el diagnóstico prenatal y el diagnóstico genético preimplantacional.
También se pueden identificar casos mediante cribados de población e investigaciones posteriores.
|
Un cribado de población es una intervención de salud pública dirigida a buscar entre población que no tiene síntomas de una enfermedad concreta, ni ha pedido ayuda médica, los individuos que tienen más riesgo de padecerla a fin de ofrecerles pruebas diagnósticas y opciones terapéuticas o preventivas. |

El cribado de población más universal en relación a las enfermedades metabólicas hereditarias es el cribado neonatal, pero desde el punto de vista epidemiológico hay que tener en cuenta que hay unas 700 enfermedades metabólicas hereditarias.
Las que reúnen características adecuadas para ser objeto de cribado neonatal son unas 25 y no hay un criterio de inclusión universalmente aceptado para todas ellas.
El cribado neonatal está dirigido a enfermedades severas tratables, pero para algunas enfermedades severas no tratables y con incidencia elevada en determinados grupos de población existen programas de cribado de heterocigotos en el período previo a la concepción o prenatal.
2. ¿Cómo se estima la frecuencia de una enfermedad en una población?
La frecuencia de una enfermedad es el número de individuos que la padecen. Este número no resulta útil si no se refiere al tamaño de la población de donde proceden los casos y momento o período de tiempo determinado.
Por ello se introducen los conceptos siguientes:
- Prevalencia: Número de casos de una enfermedad en una población en un momento determinado. Por ejemplo, número de casos de PKU que hay en España en el momento en que se hace el registro de pacientes.
- Incidencia: Número de casos nuevos de una enfermedad durante un período de tiempo. Por ejemplo, número de nuevos casos de PKU que aparecen durante un año.
- En el caso de los defectos congénitos y los trastornos genéticos, como es el caso de las enfermedades metabólicas hereditarias, suele emplearse como medida de frecuencia la Prevalencia al Nacimiento, es decir, el número de casos observados en una población de recién nacidos lo más amplia posible.
¿Cómo se estima la frecuencia de un ECM?
Si consideramos los ECM en su conjunto, su frecuencia sería el número de pacientes diagnosticados de estas enfermedades. Hay más de 700 ECM descritos, todos ellos con una incidencia muy baja, por eso están catalogados como Enfermedades Raras o Minoritarias (según la Unión Europea aquellas cuya incidencia es inferior a 5 casos por 10.000 individuos), e incluso un 80% de ellos como Enfermedades Extra-raras o muy raras (inferior a 0.0001).
La prevalencia combinada de los ECM se estima según estudios recientes en 1 en 800 recién nacidos vivos.
En general, no es útil considerarlos en su conjunto, sino individualmente.
Es difícil estimar la frecuencia de un ECM debido a su heterogeneidad clínica (distinta presentación clínica para una misma enfermedad) y a la extrema gravedad de algunos casos, que pueden morir antes de ser diagnosticados.
Un solo diagnóstico perdido hace variar mucho la frecuencia en enfermedades tan minoritarias.
Son muy útiles los datos obtenidos a partir de los programas de cribado neonatal y del cribado de portadores, cuando se dispone de ellos.
Hay que destacar que la prevalencia al nacimiento obtenida a partir de dichos programas no es un dato totalmente exacto, ya que no incluye los casos que mueren antes del cribado ni tampoco los falsos negativos, que no se detectan en la prueba.
Por ello algunos autores emplean el término "tasa de detección", es decir, el número de recién nacidos al que hay que realizar la prueba de cribado para encontrar un caso de la enfermedad.
En la tabla Frecuencia de ECM que son objeto de cribado neonatal se comparan las prevalencias al nacimiento de algunos ECM que son objeto de cribado neonatal en los países que en estos momentos tienen datos de los números más elevados de recién nacidos, Alemania (período 2005-2008) y EUA (período 2001-2006).
Se puede observar que no están incluidas las mismas enfermedades en los programas, la diferente frecuencia de algunas enfermedades en ambos países y las prevalencias extremadamente bajas de algunas.
Los registros de pacientes son también útiles para calcular la frecuencia de los ECM.
¿De qué depende la frecuencia de un ECM en una población?
Depende de la tasa de mutaciones de un gen, del patrón de herencia de la enfermedad y de las desventajas conferidas por la mutación.
Influyen también fenómenos distorsionadores naturales y sociales, como el patrón de migraciones, la consanguinidad de las parejas, la endogamia entre individuos que pertenecen a grupos étnicos o sociales restringidos o la aplicación sistemática de recursos a la prevención y al control.
¿Qué significa la tasa de mutaciones de un gen?
La tasa de mutación de un gen es la probabilidad de que se produzca una mutación en el mismo en cada generación y varía a través de las diferentes regiones del genoma.
En ausencia de sucesos distorsionadores, la frecuencia de una mutación se mantiene constante en las generaciones siguientes.
En un estudio de 1.000 genomas se ha establecido que existe una tasa de mutaciones de sustitución de bases germinales del DNA "de novo" de 10-8 por par de bases y generación.
Se ha observado también que cada persona porta de 50 a 100 variantes previamente implicadas en trastornos hereditarios.
¿Qué significa el que una mutación implique desventajas?
Cuando una mutación en un gen altera la función de una proteína, esto da lugar a una desventaja en el individuo que la padece y la versión mutante del gen tiende a desaparecer de la población.
No obstante, aparecen nuevas mutaciones en cada generación y se alcanza un equilibrio entre una mutación que da lugar a una variante deletérea (dañina, nociva) del gen y la selección natural que la elimina de la población.
Los ECM son tan minoritarios porque sus tasas de mutación por gen y generación son muy pequeñas y sus desventajas selectivas muy grandes, lo que hace que el balance entre mutación y selección sea muy bajo.
¿Existen mutaciones que impliquen ventajas?
Si implican ventajas, ya no se consideran mutaciones sino cambios de adaptación de las especies al medio ambiente.
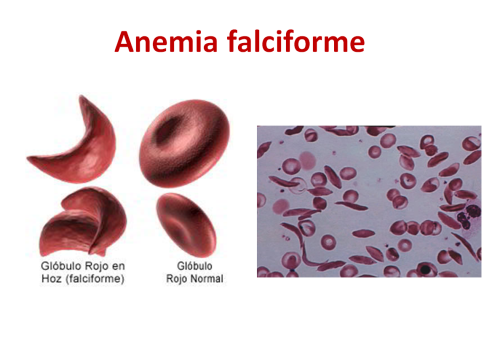
Existen, sin embargo, mutaciones en un gen que causan enfermedades autosómicas recesivas, en las que el carácter de portador (heterocigoto, es decir, con un único alelo mutado) implica ventajas en relación al homocigoto normal (dos alelos sin mutaciones) o el homocigoto con la enfermedad (dos alelos con la mutación deletérea).
Por ejemplo, la anemia falciforme, cuyos portadores muestran mayor resistencia frente a la malaria que los homocigotos anémicos para el gen mutado o los individuos normales. Esta característica o ventaja selectiva ha hecho que en 35 generaciones haya aumentado la frecuencia del gen mutado del 0,1% al 45% en África oriental.
¿Cómo influye el patrón de migraciones?
El patrón de migraciones es la tendencia a migrar (desplazarse) a otros países por diversas causas como invasiones bélicas históricas y movimientos sociales.
Este patrón es determinante en la extensión de las enfermedades hereditarias a nuevas áreas geográficas (ver también efecto fundador).
¿Y la consanguinidad?
Se produce cuando se unen individuos pertenecientes a una misma familia. Influye especialmente en las enfermedades que se heredan de forma autosómica recesiva, ya que al ser consanguíneos los padres es más probable que compartan mutaciones poco frecuentes, lo que aumenta la probabilidad que las transmitan a la descendencia.
¿Qué es la endogamia?
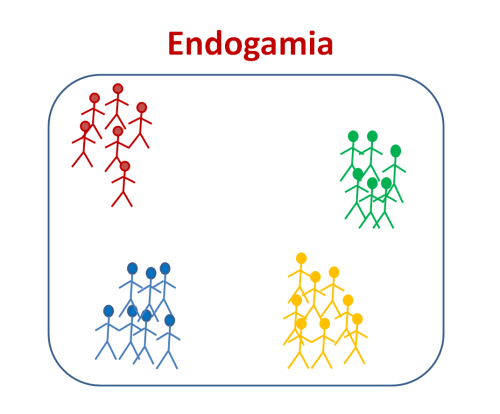
Es la unión entre individuos de ascendencia común, de una misma familia o linaje, habitualmente con objeto de garantizar la unidad de la tribu o clan.
Aumenta la probabilidad de que los cónyuges compartan la misma información genética, con lo que la frecuencia de ECM de herencia autosómica recesiva aumenta considerablemente.
¿Existen otras causas de aumento de frecuencia de un ECM en una población?
Otra causa es el efecto fundador, que se produce cuando una población pasa de una rápida reducción de tamaño a una rápida extensión del mismo. Las mutaciones presentes en la población empequeñecida de origen, se mantienen en cuando ésta se expande y dan lugar a un aumento de la frecuencia aparente.
¿Se puede reducir la frecuencia de los ECM en una población?
Es difícil dados los mecanismos biológicos que los causan, pero sí que pueden tomarse medidas de prevención y control. Para ello es imprescindible el conocimiento de la historia natural de la enfermedad, es decir la descripción de la progresión esperada de la enfermedad en el tiempo en un grupo de pacientes suficientemente representativo, desde el diagnóstico o debut de la enfermedad hasta el fallecimiento, es decir desde el inicio biológico, pasando por la fase preclínica, a la fase clínica y el desenlace.
Este conocimiento de la historia natural de la enfermedad es necesario para poder evaluar los resultados de las intervenciones dirigidas a modificarla positivamente y los beneficios conseguidos y también para diseñar dichas intervenciones, porque las medidas a tomar difieren significativamente según las fases de la historia natural.
Distinguiremos así:
- La prevención primaria: dirigida a reducir la frecuencia de una enfermedad reduciendo el número de nuevos casos que aparecen en una población definida. Las opciones parten del ofrecimiento de consejo genético a los padres, proceso que incluye las opciones preventivas a nivel familiar como, la detección de portadores, el diagnóstico prenatal y los recursos que brindan las tecnologías de reproducción asistida.
Esta aproximación está dirigida a la unidad familiar e implican el conocimiento del riesgo genético cuando ya se ha diagnosticado un enfermo, pero existe una aproximación más anticipativa a nivel de la población general, que se aplica cuando la frecuencia de una enfermedad severa y sin tratamiento es elevada en una población concreta.
Se trata del cribado de población de heterocigotos en adultos jóvenes en el período pre-concepcional o prenatal. Estos programas han reducido en un 90% la presencia de la enfermedad de Tay-Sachs en comunidades asquenazitas y en Cerdeña han reducido en un 90% los casos de talasemia major que han pasado de 1 en 250 nacimientos a 1 en 4.000.
- La prevención secundaria: dirigida a reducir la prevalencia, reduciendo la duración del trastorno o de las disfunciones asociadas al mismo, mediante la intervención precoz y tratamiento en individuos que han expresado signos y síntomas.
Para algunos ECM, como la fenilcetonuria, el éxito del tratamiento depende del hecho de que se detecte la enfermedad antes de la sospecha clínica en base a los síntomas, siendo este el origen de los programas de cribado neonatal.
Los ECM son enfermedades genéticas y por lo tanto crónicas, los programas de cribado neonatal no se ha demostrado por el momento que cambien substancialmente la prevalencia, pero sí que mejoran extraordinariamente la calidad de vida y la supervivencia de los afectados por lo que son considerados valiosas intervenciones de salud pública en forma de prevención secundaria.
Las intervenciones terapéuticas en pacientes que han expresado síntomas de ECM que por diversas razones no son objeto del cribado neonatal, que son la inmensa mayoría de ECM, también han registrado importantes progresos, pero faltan datos epidemiológicos rigurosos y comparables para conocer y cuantificar la efectividad de las medidas de prevención secundaria, en general.
- La prevención terciaria: dirigida a reducir las discapacidades y la dependencia, y a prevenir los hándicaps a pesar de la persistencia de la alteración. Junto con el tratamiento, la prevención terciaria incluirá los aspectos psicosociales, educativos, familiares y sociales. Es necesario establecer las necesidades de servicios para las personas con ECM a fin de proporcionarles unos servicios integrales y equitativos.
Documentos
Resumen del Discurso de Ingreso de la Dra. Teresa Pàmpols como Académica Numeraria de la Reial Acadèmia de Farmàcia de Catalunya. Notas tomadas por la Dra. Vilaseca.
- 12533 lecturas
También te puede interesar

 y el Registro Unificado Europeo para Enfermedades Metabólicas Hereditarias (U-IMD)")