Errores innatos relacionados con el metabolismo del triptófano
Resumen de la Cápsula Metabólica presentada por la Dra. Clara Oliva, en la Unidad de Enfermedades Metabólicas del Hospital Sant Joan de Déu Barcelona el 25 de febrero de 2022.

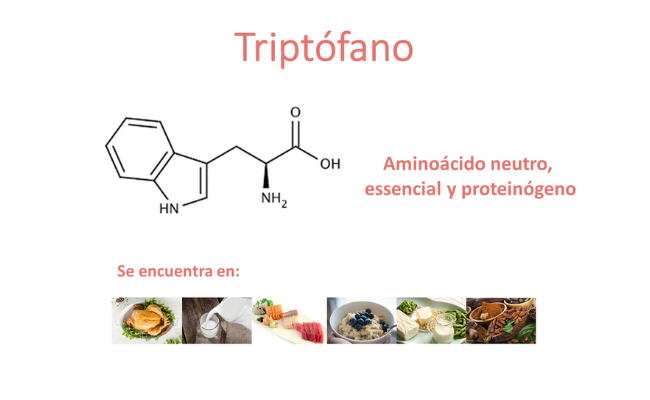
El triptófano es un aminoácido neutro, esencial (no es generado por nuestro cuerpo y debe ingerirse con la dieta) y proteinógeno (que forma parte de las proteínas). Se encuentra naturalmente en las aves, leche, soja, cereales, chocolate, plátano, piña, pescado, huevos…
¿Cómo se metaboliza el triptófano?
El triptófano se metaboliza por tres vías diferentes:
1.- La vía de la kinurenina, precursora de la niacina (vitamina B3), que interviene en la síntesis y reciclaje de NAD+. El NAD+ y NADH (su forma reducida) están implicados en las reacciones de reducción-oxidación. Los defectos de síntesis y reciclaje de NAD+ (NAXD (#618321), NADK2D, OMIM (#616034) y NAXE (#617186) se pueden considerar relacionados con el metabolismo del triptófano.
En la vía de la kinurenina propiamente dicha se han descrito la triptofanuria y la deficiencia de kinureninasa, aunque no son defectos relevantes.
Por otra parte, la kinurenina es un precursor del ácido glutárico, por lo que el triptófano inicialmente se había restringido en la dieta especial de los pacientes con aciduria glutárica (actualmente se ha observado que esta medida no es efectiva y se ha abandonado).
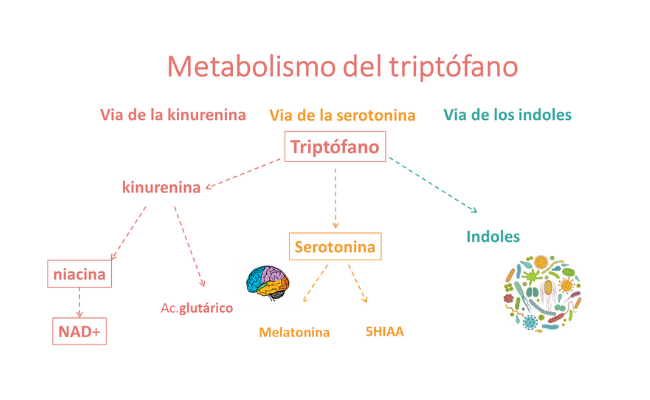
2.- La vía de la serotonina (5-hidroxitriptamina), neurotransmisor cerebral, cuya deficiencia puede resultar en fenotipos conductuales y psiquiátricos. Entre los defectos relacionados con el metabolismo de la serotonina se hallan los defectos del metabolismo de las pterinas, la deficiencia de la decarboxilasa de aminoácidos y los defectos de la vitamina B6. La melatonina es un metabolito de la serotonina, que regula los ciclos de sueño-vigilia.
3.- La vía de los indoles es la ruta principal del metabolismo del triptófano en el microbiota, que parece que tiene impacto en el sistema inmunitario.
¿Cómo se transporta el triptófano en el intestino y el riñón?
El triptófano se absorbe en el intestino y en el riñón mediante el transportador de aminoácidos neutros B0AT1, codificado por el gen SLC6A19. La proteína transportadora B0AT1 se expresa en el intestino y el riñón, y la pérdida de su función debida a alteraciones genéticas (mutaciones) conduce a la enfermedad de Hartnup, que cursa con una absorción deficiente de aminoácidos neutros que se excretan en las heces y la orina.
Además de la aminoaciduria, la pérdida de estos aminoácidos neutros puede provocar deficiencias en los metabolitos precursores, que son responsables de otros fenotipos asociados con la enfermedad de Hartnup.
El triptófano es un precursor del niacina, cuya deficiencia provoca la pelagra (dermatitis, diarrea, demencia). El triptófano también es un precursor de la serotonina, un neurotransmisor cerebral.
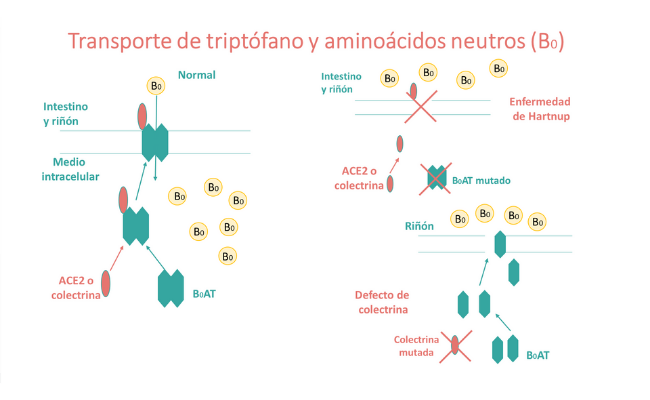
Recientemente se han descrito dos pacientes con un defecto de la proteína colectrina, que tiene una función en el tráfico del transportador B0AT1 a la superficie celular y su activación catalítica.
En el riñón, la proteína B0AT1 se une a la colectrina para su expresión superficial y estabilidad. Mutaciones en el gen CLTRN, que codifica a la colectrina causan un patrón particular de aminoaciduria, fenotipos psiquiátricos y conductuales y una dermatitis fotosensible intermitente similares a los que se observan en la enfermedad de Hartnup.
Consideraciones generales
El metabolismo del triptófano es complejo y da lugar a la síntesis de una serie de compuestos de gran importancia metabólica, como el niacina, NAD+ y serotonina.
La descripción de nuevas enfermedades metabólicas de origen genético aporta un conocimiento más profundo acerca de la enorme complejidad de las rutas metabólicas y de la función de los diferentes metabolitos y proteínas que las integran, debido a las alteraciones clínicas y metabólicas asociadas los nuevos cambios genéticos descritos. Este conocimiento puede tener consecuencias favorables en el diseño de nuevos tratamientos basados en evitar la disfunción causada por las alteraciones genéticas.
Referencia
- Pillai NR, Yubero D, Shayota BJ, Oyarzábal A, Ghosh R, Sun Q, Azamian MS, Arjona C, Brandi N, Palau F, Lalani SR, Artuch R, García-Cazorla A, Scott DA. Loss of CLTRN function produces a neuropsychiatric disorder and a biochemical phenotype that mimics Hartnup disease. Am J Med Genet A. 2019 Dec;179(12):2459-2468. doi: 10.1002/ajmg.a.61357. Epub 2019 Sep 13. PMID: 31520464.
Más información en Guía Metabólica
- 5264 lecturas
También te puede interesar


