Deficiencia de BCKDK, una enfermedad del neurodesarrollo tratable, susceptible de detección neonatal
Resumen del artículo: Tangeraas T, Constante JR, Backe PH, Oyarzábal A, Neugebauer J, Weinhold N, Boemer F, Debray FG, Ozturk-Hism B, Evren G, Tuba EF, Ummuhan O, Footitt E, Davison J, Martinez C, Bueno C, Machado I, Rodríguez-Pombo P, Al-Sannaa N, De Los Santos M, Muchart López J, Ozturkmen-Akay H, Karaca M, Tekin M, Pajares S, Ormazabal A, Stoway SD, Artuch R, Dixon M, Mørkrid L, García-Cazorla A. BCKDK deficiency: a treatable neurodevelopmental disease amenable to newborn screening. Brain. 2023 Feb 2:awad010.

La deficiencia de cetoácido deshidrogenasa quinasa de cadena ramificado (BCKDK) causa un defecto de aminoácidos de cadena ramificada (BCAA) y se asocia con un trastorno del neurodesarrollo caracterizado por autismo, discapacidad intelectual y microcefalia.
En este artículo se presenta la mayor cohorte de pacientes estudiados, ampliando el espectro fenotípico y genotípico. Además, este es el primer estudio que presenta resultados del cribado neonatal y del tratamiento clínico a medio plazo.
¿Qué es la deficiencia de BCKDK?
La cetoácido deshidrogenasa de cadena ramificada (BCKDH) es un complejo enzimático mitocondrial compuesto por cuatro subunidades catalíticas E1α, E1β, E2-DBT y E3-DLD. Está regulado por dos enzimas: la fosforilasa quinasa que lo inactiva y una fosfatasa que lo activa.
Cuando los niveles de BCAA son excesivos, el complejo BCKDH está en su forma activa y cataboliza los BCAA que, en concentraciones elevadas (especialmente la leucina), son tóxicos para el cerebro.
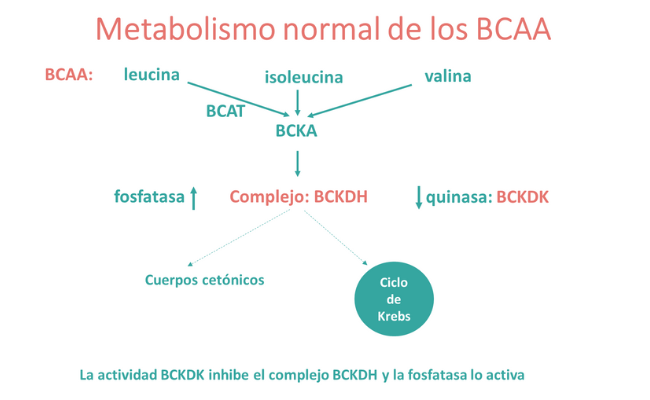
La deficiencia de BCKDH causa la enfermedad de la orina con olor a jarabe de arce (MSUD), en la que la acumulación de BCAA y sus correspondientes cetoácidos tienen un efecto tóxico sobre la función cerebral.
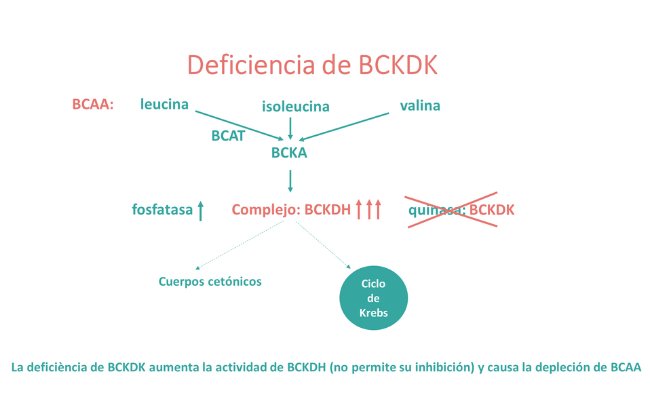
Por el contrario, cuando la BCKDH está constantemente sobreactivada debido a mutaciones en la quinasa reguladora (BCKDK), se produce una inhibición de la oxidación de BCAA que conduce a la deficiencia de estos aminoácidos esenciales, lo que afecta la síntesis de proteínas durante el desarrollo y el crecimiento posnatal (1,2).
Deficiencia de cetoácido deshidrogenasa quinasa de cadena ramificada (BCKDK)
La deficiencia de BCKDK (OMIM 614901) fue descrita por primera vez por Novarino y col. (1) en 2012 como una forma mendeliana de autismo con discapacidad intelectual y epilepsia, asociados niveles plasmáticos bajos de aminoácidos ramificados (BCAA). En 2014 se describieron dos nuevos casos con variantes genéticas nuevas y un fenotipo similar (2).
El presente texto presenta la mayor cohorte de pacientes estudiados, ampliando el espectro fenotípico y genotípico. Además, este es el primer estudio que presenta resultados del cribado neonatal y resultado clínico a medio plazo (3).
Resultados
Pacientes
Se incluyeron 21 pacientes pertenecientes a 13 familias, que presentaban bajos niveles de BCAA en plasma y líquido cefalorraquídeo y portaban mutaciones patogénicas en el gen BCKDK.
Los pacientes fueron diagnosticados entre los ocho meses y los 16 años (media: 5,8 años, 43% mujeres) y fueron reclutados a través de una iniciativa de MetabERN desde ocho países: Bélgica, Brasil, Alemania, Noruega, Arabia Saudita, España, Turquía y el Reino Unido durante 2018-2021.
Presentación clínica antes del tratamiento
Todos los pacientes tenían un retraso global en el desarrollo neurológico; 18/21 tenían deterioro de la función motora gruesa, 17/17 discapacidad intelectual, 17/17 deterioro del lenguaje, 12/17 trastorno del espectro autista, 9/21 epilepsia, 12/15 torpeza, 3/21 tenían pérdida auditiva neurosensorial y 4/20 dificultades de alimentación. No se observó microcefalia al nacer, pero 16/20 desarrollaron microcefalia durante el seguimiento. Cinco pacientes mostraban regresión. Se observó un trastorno del movimiento en 3/21 pacientes: movimientos hipercinéticos (1), ataxia troncal (1) y distonía (2).
Tratamiento
Después del tratamiento con dieta hiperproteica (≥ 2g/kg/día) y suplemento de BCAA (100-250 mg/kg/día), los BCAA en plasma aumentaron significativamente (p < 0,001), las funciones motoras y el perímetro cefálico se estabilizaron/mejoraron en 13/13 y en 8/13 pacientes, respectivamente.
Entre los casos con datos de seguimiento, ninguno de los tres pacientes que iniciaron el tratamiento antes de los dos años de edad desarrolló autismo. El paciente con la edad más temprana de inicio del tratamiento (ocho meses) mostró un desarrollo normal a los tres años de edad.
Datos de la detección neonatal de enfermedades metabólicas
La detección neonatal identificó niveles de BCAA significativamente más bajos que los de la población normal.
Discusión
Los BCAA son aminoácidos esenciales involucrados en reacciones celulares vitales como la regulación del recambio de proteínas, la señalización de la autofagia y la función mitocondrial. Además, su metabolismo está acoplado al del neurotransmisor glutamato.
La activación mantenida del complejo BCKDH, debida a mutaciones en el gen BCKDK da lugar a un hipermetabolismo de los BCAA que causa unos bajos niveles corporales de los mismos. Como la síntesis de proteínas depende de la disponibilidad de aminoácidos, la deficiencia de estas moléculas esenciales, entre otros mecanismos, afecta el neurodesarrollo.
En los tres pacientes en los que se inició tratamiento con BCAA antes de los dos años de edad, se observó una mejora del retraso en el desarrollo en comparación con los pacientes mayores.
Dado que el tratamiento adecuado desde el nacimiento puede tener el potencial de mejorar el desarrollo neurológico, se sugiere que esta condición es susceptible de detección neonatal, basada en la medición de aminoácidos, ya que seis de los siete pacientes incluidos en este estudio mostraron un perfil anormal de BCAA en la detección neonatal.
Referencias
- Novarino G, El-Fishawy P, Kayserili H, Meguid NA, Scott EM, Schroth J, Silhavy JL, Kara M, Khalil RO, Ben-Omran T, Ercan-Sencicek AG, Hashish AF, Sanders SJ, Gupta AR, Hashem HS, Matern D, Gabriel S, Sweetman L, Rahimi Y, Harris RA, State MW, Gleeson JG. Mutations in BCKD-kinase lead to a potentially treatable form of autism with epilepsy. Science. 2012 Oct 19;338(6105):394-7. doi: 10.1126/science.1224631. Epub 2012 Sep 6. PMID: 22956686; PMCID: PMC3704165.
- García-Cazorla A, Oyarzabal A, Fort J, Robles C, Castejón E, Ruiz-Sala P, Bodoy S, Merinero B, Lopez-Sala A, Dopazo J, Nunes V, Ugarte M, Artuch R, Palacín M, Rodríguez-Pombo P, Alcaide P, Navarrete R, Sanz P, Font-Llitjós M, Vilaseca MA, Ormaizabal A, Pristoupilova A, Agulló SB. Two novel mutations in the BCKDK (branched-chain keto-acid dehydrogenase kinase) gene are responsible for a neurobehavioral deficit in two pediatric unrelated patients. Hum Mutat. 2014 Apr;35(4):470-7. doi: 10.1002/humu.22513. Epub 2014 Mar 5. PMID: 24449431.
- Tangeraas T, Constante JR, Backe PH, Oyarzábal A, Neugebauer J, Weinhold N, Boemer F, Debray FG, Ozturk-Hism B, Evren G, Tuba EF, Ummuhan O, Footitt E, Davison J, Martinez C, Bueno C, Machado I, Rodríguez-Pombo P, Al-Sannaa N, De Los Santos M, Muchart López J, Ozturkmen-Akay H, Karaca M, Tekin M, Pajares S, Ormazabal A, Stoway SD, Artuch R, Dixon M, Mørkrid L, García-Cazorla A. BCKDK deficiency: a treatable neurodevelopmental disease amenable to newborn screening. Brain. 2023 Feb 2:awad010. doi: 10.1093/brain/awad010. Epub ahead of print. PMID: 36729635.
- 972 lecturas
También te puede interesar


