Defecto genético del transportador de multivitaminas codificado por el gen SLC5A6, potencialmente tratable
Resumen de la Cápsula Metabólica presentada por la Dra. Àngels García-Cazorla, en la Unidad de Enfermedades Metabólicas Congénitas del Hospital Sant Joan de Déu Barcelona el 8 de abril de 2021.

El transportador de multivitaminas codificado por el gen SLC5A6 es un transportador dependiente de sodio que facilita el paso de las vitaminas del grupo B, biotina y pantotenato, así como del lipoato. La proteína transportadora se expresa de forma ubicua y tiene un papel importante en la absorción de estas vitaminas en el sistema digestivo, así como en el transporte de las mismas, a través de la barrera hematoencefálica.
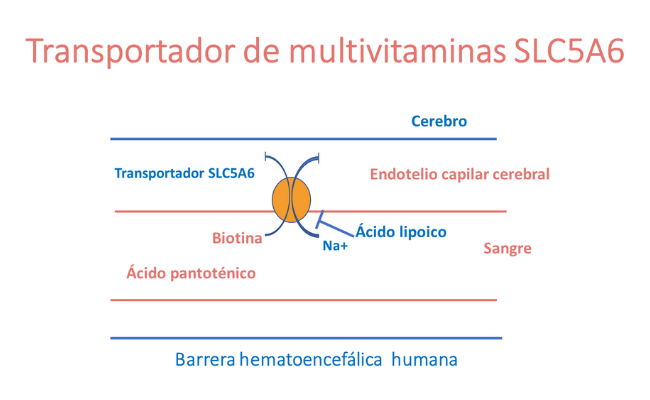
¿Qué es la biotina?
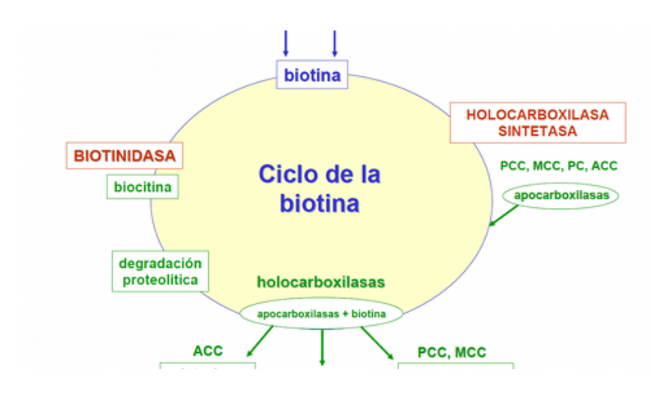
La biotina (vitamina B7) actúa como cofactor activador de las carboxilasas involucradas en una variedad de reacciones metabólicas, incluida la síntesis de ácidos grasos, gluconeogénesis y catabolismo de aminoácidos.
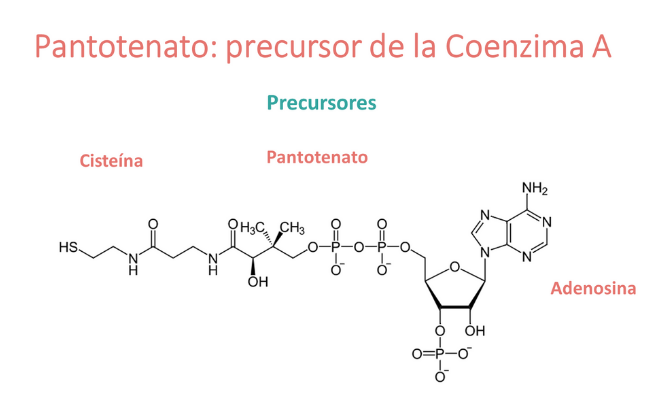
¿Qué es el pantotenato?
El ácido pantoténico (vitamina B5) es el precursor de la coenzima A y, por lo tanto, la deficiencia de ácido pantoténico puede conducir a una disponibilidad limitada de coenzima A. La coenzima A opera como cofactor de enzimas involucradas en el ciclo del ácido tricarboxílico y el metabolismo de los ácidos grasos.
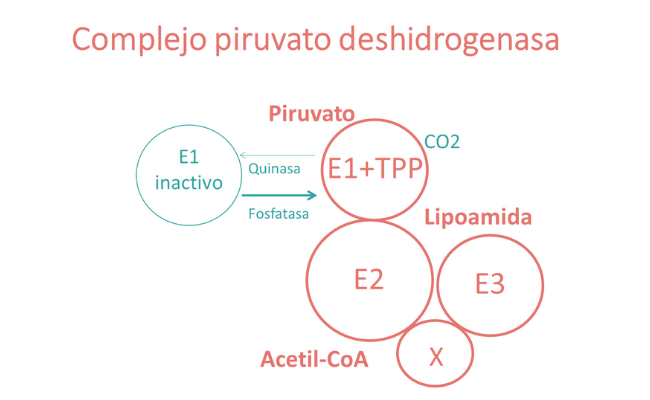
¿Qué es el lipoato?
El lipoato es uno de los cofactores en el sistema de escisión de glicina y los complejos piruvato deshidrogenasa, deshidrogenasa de los cetoácidos de cadena ramificada y cetoglutarato deshidrogenasa. Estas enzimas catalizan reacciones redox en la producción mitocondrial de energía y permiten la descarboxilación oxidativa de reacciones de aminoácidos y cetoácidos. Además de su función como cofactor, el lipoato también puede tener propiedades antioxidantes y efectos antiinflamatorios.
¿Qué efectos metabólicos tiene el defecto genético del transportador de multivitaminas?
Mutaciones en el gen SLC5A6 causantes de una proteína transportadora con una función alterada pueden dar lugar a una deficiencia intracelular de sustratos orgánicos, ya que es el único transportador combinado conocido de biotina, ácido pantoténico y lipoato. Teniendo en cuenta sus funciones metabólicas, las deficiencias intracelulares de estas vitaminas pueden causar una amplia variedad de signos y síntomas clínicos.
¿Se han descrito pacientes con mutaciones en el gen SLC5A6?
Se han descrito recientemente unos pocos pacientes con mutaciones en este gen (ver referencias).
El primer paciente, a los 15 meses de edad, mostró retraso del crecimiento, microcefalia y cambios en la resonancia magnética cerebral, parálisis cerebral y retraso en el desarrollo, inmunodeficiencia variable, reflujo gastroesofágico, osteoporosis y fracturas óseas. El estudio genético identificó mutaciones (R94X, una terminación prematura, y R123L, un cambio disfuncional de aminoácidos), en el gen SLC5A6 (Hum Genet, 2017).
La segunda paciente, una niña de 17 meses, presentó hipoglucemia y acidosis metabólica grave, que requirió reanimación. Su historia reveló problemas de alimentación desde el nacimiento y poco aumento de peso. La investigación metabólica mostró niveles plasmáticos elevados de C3-carnitina y C5-OH-carnitina. El análisis de orina mostró una excreción persistentemente elevada de ácido 3-OH-isovalérico, todo ello compatible con una deficiencia de biotinidasa, que no pudo demostrarse. La secuenciación completa del exoma reveló heterocigosidad compuesta para variantes en el gen SLC5A6 (JIMD Rep, 2019).
Simultáneamente se describieron dos hermanos con una presentación de inicio infantil (12 a 14 meses), con un fenotipo neurodegenerativo progresivo, retraso del desarrollo y encefalopatía epiléptica. Uno de los hermanos falleció debido a una hemorragia gastrointestinal por perforación de una úlcera. La secuenciación completa del exoma reveló mutaciones en el gen SLC5A6 (NPJ Genom Med, 2019).
¿Tiene tratamiento este defecto?
Al confirmarse el diagnóstico genético de mutaciones en SLC5A6, estos pacientes fueron tratados con biotina, pantotenato y ácido lipoico. El tratamiento mejoró las funciones neurocognitivas y neuromotores de estos pacientes.
Conclusiones
- Las manifestaciones clínicas de estos pacientes (todos ello con manifestaciones neurológicas) sugirieron la conveniencia de proseguir el estudio metabólico, que sólo en el segundo caso fue informativo. Únicamente la secuenciación completa del exoma aportó el diagnóstico definitivo de estos pacientes.
- Por otra parte, el diagnóstico de estos pacientes sugiere que muchas de las enfermedades metabólicas diagnosticadas recientemente pueden no mostrar marcadores bioquímicos orientativos del defecto, por no ser causados directamente por bloqueos totales en vías metabólicas sino por alteraciones metabólicas más sutiles, como los defectos de transporte de cofactores.
- Esto demuestra la importancia del estudio genético ante posibles enfermedades metabólicas, potencialmente tratables.
Referencias
- Subramanian VS, Constantinescu AR, Benke PJ, Said HM. Mutations in SLC5A6 associated with brain, immune, bone, and intestinal dysfunction in a young child. Hum Genet 2017;136(2):253-261.
- Schwantje M, de Sain-van der Velden M, Jans J, van Gassen K, Dorrepaal C, Koop K, Visser G. Genetic defect of the sodium-dependent multivitamin transporter: A treatable disease, mimicking biotinidase deficiency. JIMD Rep. 2019;48(1):11-14.
- Byrne AB, Arts P, Polyak SW, Feng J, Schreiber AW, Kassahn KS, Hahn CN, Mordaunt DA, Fletcher JM, Lipsett J, Bratkovic D, Booker GW, Smith NJ, Scott HS. Identification and targeted management of a neurodegenerative disorder caused by biallelic mutations in SLC5A6. NPJ Genom Med 2019;4:28.
- 517 lecturas
También te puede interesar


