Se presenta en el ICIEM Barcelona el descubrimiento de diez nuevos Errores Congénitos del Metabolismo

En el pasado congreso ICIEM celebrado en Barcelona en septiembre de este año se han descrito 10 nuevas enfermedades metabólicas hereditarias, hecho que tiene una enorme importancia para avanzar en el disgnóstico y tratamiento de pacientes hasta ahora no detectados.
En primer lugar, permite diagnosticar a más pacientes que las padecen y que han permanecido sin diagnóstico específico hasta el momento de la descripción detallada del origen bioquímico-molecular de las mismas. Este diagnóstico puede representar para los pacientes la posibilidad de un tratamiento, y para sus familias una mayor información sobre el curso de la enfermedad y el pronóstico.
Asimismo, pueden disponer de un consejo genético más preciso y la posibilidad, en algunos casos, de diagnóstico prenatal, si se requiere.
En segundo lugar, el descubrimiento de una nueva enfermedad representa un conocimiento mejor de los eslabones de una vía metabólica, que hasta el momento se desconocían, así como de las funciones específicas de cada uno de los elementos que intervienen en la proteína afectada por las mutaciones génicas, conociendo así su función determinada en el metabolismo humano.
Entre los nuevos ECM descritos, cuatro están relacionados con el metabolismo energético mitocondrial, tres pertenecen al metabolismo intermediario, uno es un defecto de transporte, otro es un nuevo defecto congénito de la glicosilación (CDG) y, finalmente, se describe un defecto de un canal de sodio.
Metabolismo energético mitocondrial
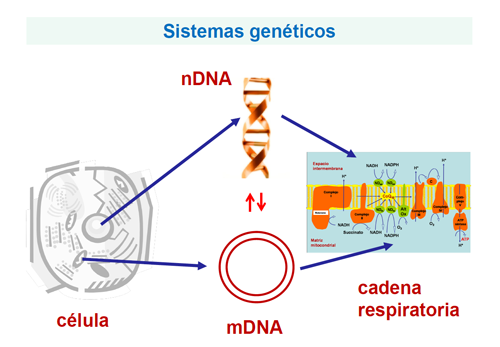
Las cuatro enfermedades del metabolismo energético mitocondrial son defectos en proteínas de la cadena respiratoria (CR) codificadas por el DNA nuclear (nDNA).
Los defectos de la CR mitocondrial engloban un grupo de trastornos bien definidos desde el punto de vista bioquímico, pero muy heterogéneos desde el punto de vista clínico y particularmente genético, muchos de los cuales aún permanecen sin diagnóstico genético.
Esto se debe a que se desconocen en detalle las proteínas y genes implicados en un proceso biológico tan esencial como es la cadena de transporte electrónico mitocondrial, que conduce a la producción de energía para todos los procesos metabólicos.
- Mutación en homocigosis en el gen IBA57 que causa un defecto de la biosíntesis de los clústeres hierro-azufre, que afectan a los complejos I y II de la cadena respiratoria mitocondrial. Este defecto se ha descrito en dos hermanos con una encefalomiopatía(afectación cerebral y muscular) neonatal fatal (Vanlander y col, Bélgica).
- Mutaciones en el gen FBX14 que causan depleción (disminución en el número de copias del DNA mitocondrial) del DNA mitocondrial (mtDNA) al afectar a la integridad y estabilidad del mismo. Este defecto se ha descrito en tres familias consanguíneas no relacionadas en las que los niños afectos presentaron una encefalopatía fatal, con depleción del mtDNA y deficiencia grave de la cadena respiratoria (Bonnen y col, Houston EEUU, RU y Arabia Saudí).
- Mutaciones en el gen ELAC2 que causan un defecto en la transferencia mitocondrial de precursores de RNA, asociado a cardiomiopatía hipertrófica y retraso psicomotor de inicio y gravedad variables. Este defecto se ha descrito en 5 pacientes de 3 familias, que presentaban acidosis láctica y un defecto de los complejos I ó I/IV de la cadena respiratoria. El hallazgo de estas mutaciones génicas permite asociar por primera vez el procesamiento de RNA con una enfermedad humana (Freisinger y col, Alemania, Italia, RU y Austria).
- Mutaciones en el gen LYRM4, que codifican para el factor ISD11 implicado en la biogénesis de los clústeres hierro-azufre, causan una deficiencia múltiple de los complejos de la CR mitocondrial.
Se ha descrito en un paciente y su primo fallecido en el período neonatal, ambos con deficiencia de los complejos I, II y III de la cadena respiratoria, así como de otras enzimas (aconitasa y ferroquelatasa), que contienen clústeres hierro-azufre.
La diferente presentación clínica en ambos pacientes sugiere la importancia de la biodisponibilidad (disponibilidad para el organismo) del aminoácido cisteína en el período neonatal y da pistas para un posible tratamiento de esta enfermedad (Thornburn y col, Australia, Alemania, EEUU).
Metabolismo intermediario
- Mutaciones en el gen HCFC1, que codifica a un co-regulador transcripcional, causan un nuevo trastorno ligado al cromosomaX en el metabolismo intramitocondrial de la cobalamina (CBLX) con una presentación clínica grave, asociada a homocistinuria con acidemia metilmalónica.
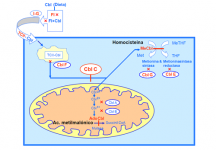
Algunos pacientes varones con dicha alteración bioquímica no mostraban mutaciones en el gen MMACHC causante de la variante CBLC y se ha demostrado que portaban una mutación con pérdida de sentido (missense) en el co-regulador transcripcional HCFC1.
Todos ellos presentaban un fenotipo clínico (grupo de manifestaciones, sintomatología) grave, con epilepsia refractaria y alteración neurocognitiva profunda, mientras que sus alteraciones bioquímicas eran más leves que las de los pacientes con CBLC.
La relación funcional entre este regulador de la transcripción global y el metabolismo de la cobalamina representa el primer ECM causado por disregulación transcripcional (Sloan y col, EEUU, Canadá y Suiza).- Mutaciones en el gen BCKDK, que inactivan la kinasa que causa una regulación negativa del complejo enzimático BCKDH. Se ha descrito este defecto en dos pacientes no relacionados con retraso grave del desarrollo, autismo, alteración del EEG y muy bajos niveles de los aminoácidos ramificados (leucina, isoleucina y valina).
Los pacientes fueron tratados con una dieta rica en proteínas y suplementada en aminoácidos ramificados, que mejoraron su perfil de aminoácidos, su crecimiento y comportamiento, demostrándose así el descubrimiento de un ECM potencialmente tratable (García-Cazorla y col, España). - Mutaciones en el gen LIPT1 como causa de un defecto de lipoilación que afecta a los complejos piruvato deshidrogenasa y α-cetoglutarato deshidrogenasa, cuyo cofactor es el ácido lipoico.
Este defecto se ha descrito en un paciente con una acidosis láctica neonatal fatal, con elevación de α-cetoglutarato y α-alanina. Esto evidencia que la alteración de la transferencia del ácido lipoico está asociada a una enfermedad en el hombre (Tort y col, España y Francia). A este trabajo se le ha otorgado el premio ICIEM 2013.
Defecto del transporte
Mutaciones en el gen SLC25A1 causan un defecto del transportador mitocondrial de citrato (CIC), que da lugar a una deficiencia combinada de aciduria D-2 y L-2-hidroxiglutárica.
Estas mutaciones se han descrito en 12 pacientes con un cuadro clínico grave asociado a estas acidurias orgánicas combinadas, cuyo origen genético se desconocía hasta ahora.
Estos autores identifican así defectos en el transportador mitocondrial de citrato SLC25A1 como nueva causa de esta variante de aciduria 2-hidroxiglutárica (Nota y col, Holanda, Francia, Qatar, Alemania, EEUU, Noruega, Suiza, Italia, Bélgica).
Defecto de la glicosilación (CDG)
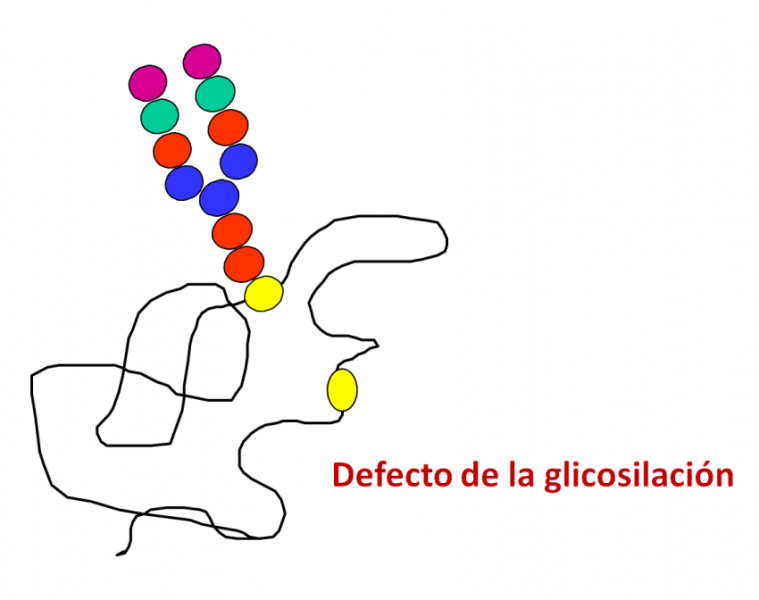
Se ha descrito un nuevo defecto congénito de la glicosilación (CDG-I), causado por mutaciones en el gen SSR3 que codifica para el receptor de señal de secuencia SSR3, asociado a la translocación de proteínas recién sintetizadas al retículo endoplasmático.
Este defecto se ha descrito en un paciente con diarrea congénita, retraso psicomotor y grave enfermedad pulmonar. Se ha demostrado que el receptor SSR3 es esencial para una eficiente N-glicosilación de las proteínas en distintos puntos (Rust y col, Alemania, Japón).
Defecto del canal de sodio
Se han descrito mutaciones en el gen NALCN, que codifica para el canal de sodio en las membranas neuronales.
Causan un síndrome autosómico recesivo descrito en dos familias con un grave fenotipo que implica dismorfia facial, hipotonía, retraso del habla, estreñimiento crónico y discapacidad intelectual.
Dado que este canal se activa por acetilcolina, esto sugiere una futura aproximación terapéutica a través de la modulación del canal (Al-Sayed y col, Arabia Saudí).
Equipo Guía Metabólica, con la colaboración de las Dras. Antònia Ribes y Judith García- Villoria del Hospital Clínic, Barcelona
- 796 lecturas
Noticias relacionadas
")
También te puede interesar


