SSIEM 2011: Avances en el diagnóstico, evolución y tratamiento de los ECM

Durante los pasados días 30 de Agosto al 3 de Septiembre tuvo lugar en Ginebra (Suiza) la Reunión Anual de la Sociedad para el Estudio de los Errores Congénitos del Metabolismo (ECM) (Annual Symposium of the Society for the Study of Inborn Errors of Metabolism, SSIEM).
En este simposium se expusieron los avances que han logrado los grupos más relevantes en el ámbito de los ECM, relacionados con el diagnóstico, evolución y/o tratamiento de diversos ECM.
En esta ocasión se otorgó gran importancia a diversos temas como:
- ECM durante la edad adulta (Adult Metabolic Workshop y Plenary Session)
- Diagnóstico precoz mediante el cribado neonatal (Newborn screening and IEM, Plenary Session)
- ECM con respuesta a vitaminas (Vitamin responsive disorders, Plenary Session)
- Defectos del metabolismo de la creatina (Creatine metabolism and related disorders, Plenary Session)
- Terapia génica (Gene therapy of IEM, Plenary Session)
ECM durante la edad adulta
Durante el taller de ECM en los adultos se expusieron diferentes casos de alteraciones metabólicas relacionadas con hipocolesterolemia que habían sido diagnosticadas durante la edad adulta (20-60 años), analizando la clínica y el perfil analítico de entidades como la Enfermedad de Tangier, la Abetalipoproteinemia, el Déficit de Lecitin colesterol acil transferasa y el Déficit de esfingomielinasa.
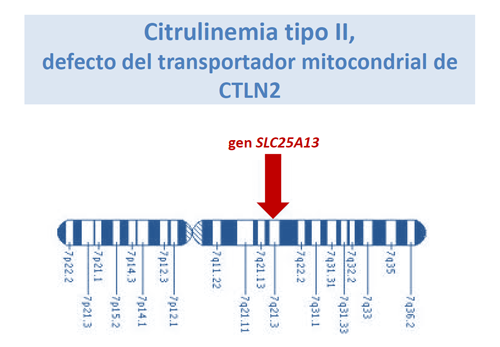
Se habló también de la Citrulinemia tipo II, causada por mutaciones en el gen SLC25A13, que dan lugar a un defecto de función de la proteína transportadora mitocondrial CTLN2.
ECM con respuesta a vitaminas
En lo referente a enfermedades con respuesta a vitaminas se consideraron aquellas relacionadas con el metabolismo de la cobalamina.
En esta charla el Dr. B Fowler explicó el reciente hallazgo de un transportador a nivel lisosomal de la cobalamina que daría lugar a unas características clínicas similares a la variante CBL-F, pero cuyo defecto se ha denominado CBL-J.
La Dra. B Plecko expuso una interesante revisión de las enfermedades con respuesta a la Vitamina B6, tiamina y biotina. Explicó el espectro de enfermedades con respuesta a la Vitamina B6, tratándose del déficit de antiquitina, el déficit de PNPO, la Infantile tissue non-specific alkaline phosphatase deficiency y el Síndrome de Mabry.

En cuanto a la biotina y la tiamina, la Dra. Plecko hizo referencia a la 3-metilcrotonilglicinuria en sus formas sensibles a la biotina, así como la Enfermedad de los Ganglios basales con respuesta a la Biotina, que más tarde se descubriría que se trata de un defecto en el transportador de tiamina tipo 2.
También el Dr. D Rosenblatt expuso con acierto el espectro de posibilidades de enfermedades con respuesta al tratamiento con ácido fólico o folínico, haciendo una revisión de las manifestaciones clínicas en la malabsorción hereditaria de ácido fólico, el déficit de MTHFR, el defecto de metionina sintasa (CBL-E/G), el defecto de folato a nivel cerebral con mutaciones en el FOLR1 o el déficit de dihidrofolato reductasa y el defecto de metilentetrahidrofolato deshidrogenasa.
En cuanto a los defectos en el metabolismo de la creatina se habló de las diferentes manifestaciones a nivel cerebral en el modelo animal del defecto de AGAT y de GAMT, observándose diferencias en cuanto a volumen de las diferentes estructuras cerebrales, como el cuerpo calloso, el hipotálamo, el tálamo y la corteza cerebral. También se apuntó que los defectos de creatina en los modelos animales pudieran asociar un defecto en la función de la cadena respiratoria mitocondrial a nivel muscular.
Se describió el segundo paciente con deficiencia de la Fosfolipasa C beta 1, que se presentó con una encefalopatía epiléptica de inicio en el primer año y, como signo clave, un pico de mioinositol en la espectroscopía (Técnica de la Resonancia Magnética) marcadamente disminuido en el primer año de vida junto con una clara atrofia cerebral.
Terapia génica
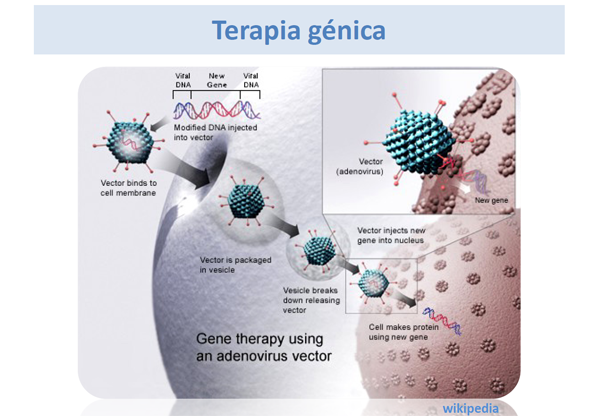
El grupo de la Dra. L. Desviat de Cantoblanco, Madrid, presentó sus estudios sobre Terapia génica en la acidemia propiónica utilizando gentamicina (antibiótico de la familia de los aminoglicósidos) en fibroblastos cultivados de los pacientes afectos, con resultados alentadores.
El grupo del Dr. Ferriero de Nápoles, Italia, presentó sus estudios en fibroblastos de pacientes con Déficit de Piruvato deshidrogenasa bajo tratamiento con fenilbutirato. Sus estudios, de forma similar a lo que se demostró con la Enfermedad de la orina con olor a jarabe de arce y la deshidrogenasa de los alfa-cetoácidos, apuntan a que el fenilbutirato es capaz de aumentar la actividad enzimática de la enzima Piruvato deshidrogenasa mediante cambios en la fosforilación de la proteína.
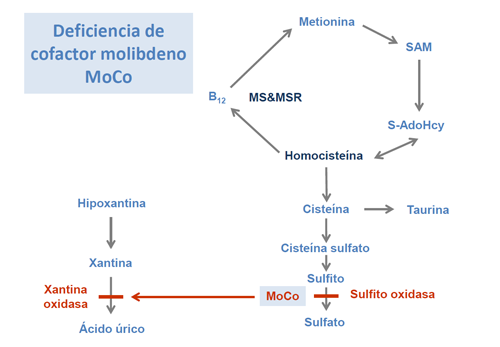
Otro noticia importante es la posibilidad de tratamiento para la deficiencia del cofactor de molibdeno (MoCo) tipo A con monofosfato de piranopterina cíclica purificada (cPMP). Este tratamiento se ha introducido ya en Guía metabólica en el apartado correspondiente a esta enfermedad, y se ha resumido el artículo que recoge esta investigación (Pediatrics 2010;125(5):e1249-54).
Otros temas de gran interés dignos de resaltar, que podrán ser tratados con mayor extensión en el apartado de Avances tecnológicos y médicos son:
- Selenio en relación con mutaciones en SBP2
- La Hipermanganesemia
- El uso de Resveratrol en el tratamiento de las enfermedades mitocondriales
- La descripción de una nueva Forma de Leucodistrofia sensible a biotina
Finalmente, hay que destacar que el equipo del Hospital Clínic formado por A. Navarro-Sastre, F. Tort, A. Font, J. García-Villoria, A. Arias, P. Alcalá, S. Moliner, C. Ogg, P. Briones y A. Ribes, de la Sección de Errores Congénitos del Metabolismo (IBC) del Servicio de Bioquímica y Genética Molecular, obtuvo el premio a la mejor comunicación.
Esta es la primera vez que este prestigioso galardón se otorga a un equipo nacional y lo ha merecido la comunicación "Tratamiento y control de los pacientes con fenilcetonuria: resultados del Grupo Colaborativo de Unidades de Seguimiento en España". En Guía metabólica os ofrecemos un resumen de dicha comunicación.
- 538 lecturas
También te puede interesar


