Tratamiento de las enfermedades metabólicas hereditarias con chaperonas farmacológicas (V/V)

Las chaperonas farmacológicas constituyen una de las nuevas aproximaciones terapéuticas de futuro dentro de la Medicina genómica. Se basan en el uso de pequeñas moléculas que ayudan a las proteínas mutantes a adoptar la forma adecuada para realizar su función de forma correcta.
Las chaperonas presentan grandes ventajas en relación con otros tratamientos usados tradicionalmente en las enfermedades metabólicas.
Opciones terapéuticas en enfermedades metabólicas hereditarias
Basándonos en su forma de actuación, podemos agruparlas en 4 tipos generales:
- Corrección metabólica
- Terapia celular
- Terapia génica
- Terapia genómica
¿En qué consiste la corrección metabólica?
La alteración metabólica, causada por la/s mutación/es en un gen, que tiene consecuencias patogénicas puede corregirse de diversas formas:
- Restricción del sustrato acumulado debido al bloqueo metabólico. Puede ir acompañada del reemplazamiento del producto no formado, que puede ser esencial. Por ejemplo: dietas especiales bajas en proteínas en muchas aminoacidopatías y acidurias orgánicas, o bien dieta libre de galactosa en la galactosemia o libre de fructosa en la fructosemia hereditaria.
- Terapia de sustitución enzimática, que consiste en administrar periódicamente una enzima activa a un paciente con una deficiencia enzimática con objeto de sustituir la deficitaria. Es un tratamiento muy empleado en enfermedades lisosomales.
- Suplemento del cofactor, que consiste en administrar el cofactor (a menudo un derivado vitamínico que optimiza la actividad enzimática) a un paciente con una deficiencia de una enzima para aumentar la actividad enzimática de la misma. Por ejemplo: defectos del metabolismo de la vitamina B12 causantes de acidurias metilmalónicas con o sin homocistinuria.
- Trasplante de órganos, para sustituir en parte la enzima deficiente, cuya actividad se expresa por ejemplo en hígado, por la enzima normal del donante. Se utiliza, por ejemplo, en defectos del ciclo de la urea y tirosinemia tipo I (hígado). El trasplante de médula ósea se utiliza en algunas enfermedades lisosomales.
- Antioxidantes, ya que se ha demostrado el importante papel del estrés oxidativo en muchas enfermedades metabólicas, por ejemplo, las enfermedades del metabolismo energético mitocondrial.
¿Qué es la terapia celular?
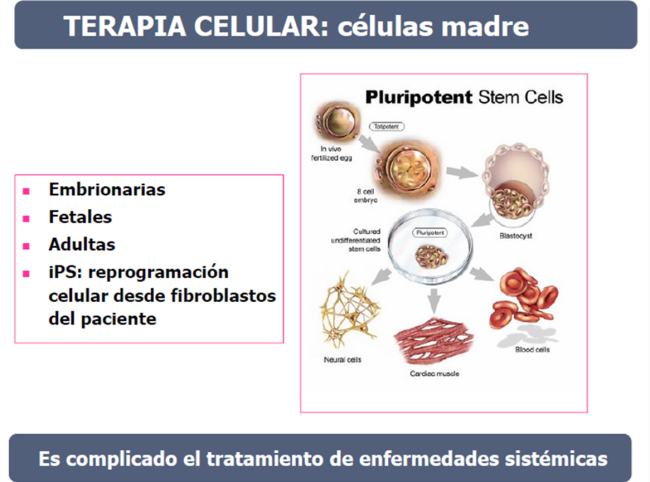
Se basa en la utilización de células madre. Su utilización es aún incipiente debido a dificultades técnicas y problemas éticos.
Estas células madre pueden ser obtenidas de cordón o médula ósea pero actualmente se está progresando en la generación de iPS (células pluripotenciales reprogramadas a partir de fibroblastos del paciente) y su diferenciación a células específicas para corregir el defecto metabólico, aunque está todavía en fase de experimentación.
¿Y la terapia génica?
La terápia génica consiste en incorporar material genético externo al material genético de una célula para modificar la acción de un gen mutado (alterado).
¿En qué consiste la terapia genómica?
Las terapias genómicas son las nuevas aproximaciones terapéuticas usadas en Medicina genómica, que se adecúa a la filosofía de la medicina actual caracterizada por las 4P: predictiva, personalizada, preventiva y participativa.
Entre ellas se hallan las terapias específicas de mutación, en las que:
- El tratamiento es específico del tipo de mutación.
- Los pacientes deben ser analizados genéticamente.
- Las mutaciones deben ser analizadas funcionalmente
¿El tipo de mutación de un gen tiene consecuencias en la proteína a la que codifica?
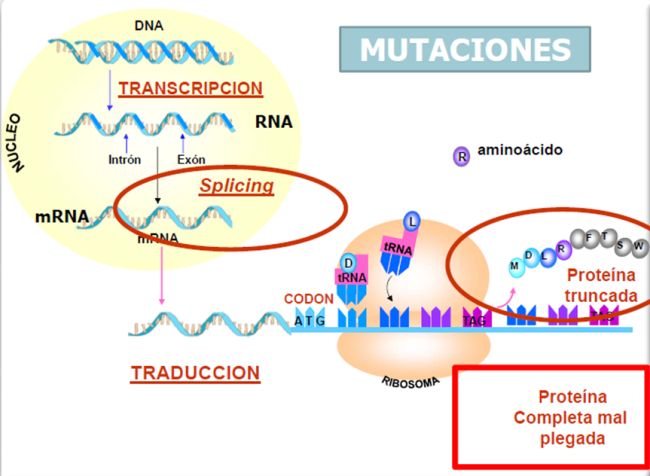
Las mutaciones pueden afectar a las proteínas a las que codifican de diferentes formas.
Por ejemplo, si interfieren en el splicing del RNA mensajero pueden causar graves cambios en la proteína mutada. También las mutaciones en las que el cambio de una base del DNA da lugar a un codón de stop prematuro (nonsense) determinan una proteína truncada.
En cambio, entre las mutaciones de cambio de base del DNA que determinan un aminoácido incorrecto en la proteína mutada (missense), se puede alterar más o menos la función de la misma. En muchos casos se afecta el plegamiento de la proteína mutada, lo que reduce su estabilidad y hace que sea fácilmente degradada y eliminada, causando así su deficiencia funcional.
¿Existen terapias específicas según el tipo de mutación?
Actualmente se investiga para fabricar terapias específicas según el tipo de mutación. Por ejemplo, para las mutaciones de splicing se investiga en terapias antisentido, para las causantes de proteínas truncadas, mediante terapias "stop codon readthrough y para las mutaciones missense, mediante la terapia con chaperonas farmacológicas.
¿Qué son las chaperonas farmacológicas?
Son pequeñas moléculas que ayudan a plegar a las proteínas mutantes de forma específica. Funcionan para las proteínas mutantes causadas por un cambio de aminoácido, que afecta a su correcto plegamiento.
¿Cuáles son las ventajas del tratamiento con chaperonas?
Tienen diversas ventajas:
- Son muchos los pacientes que se pueden tratar.
- Atraviesan la barrera hematoencefálica (a diferencia de la terapia de sustitución enzimática) o algunos cofactores, como la BH4.
- Alcanzan los diversos tejidos en donde se expresa la proteína mutada.
- Son fácilmente administrables, a diferencia de la terapia de sustitución enzimática o la terapia génica.
- No se manipula la dotación genética, a diferencia de la terapia génica.
¿Qué hacen las chaperonas?
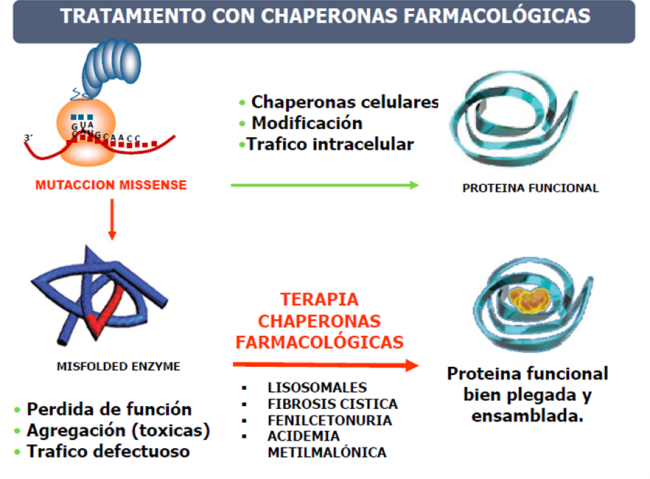
Las chaperonas celulares ayudan al plegamiento y ensamblaje correcto de las proteínas.
En las proteínas mutadas por cambio de aminoácido el plegamiento y el ensamblaje son defectuosos, se agregan fácilmente (lo cual tiene un efecto tóxico), se degradan más fácilmente o su tráfico intracelular es defectuoso.
Al ser tratadas con chaperonas farmacológicas, dichas proteínas se pliegan y ensamblan correctamente y se vuelven funcionales.
Aún cuando no se corrige toda la proteína, ésta lo hace parcialmente, aumentando en parte su función.
¿Qué enfermedades son tratables con chaperonas?
Las enfermedades causadas por proteínas mutantes que muestran:
- Un fallo de plegamiento con pérdida de función (Síndrome de Marfan, PKU, enfermedades lisosomales, etc…).
- No son suficientemente estables para realizar su función (muchas formas de cáncer).
- Un fallo en el tráfico intracelular (fibrosis quística, hipercolesterolemia familiar, deficiencia de α-1-antitripsina).
- Las que forman agregados insolubles tóxicos (enfermedades neurodegenerativas: Alzheimer, diabetes tipo II, Parkinson, etc…).
¿Cómo se descubren las chaperonas farmacológicas?
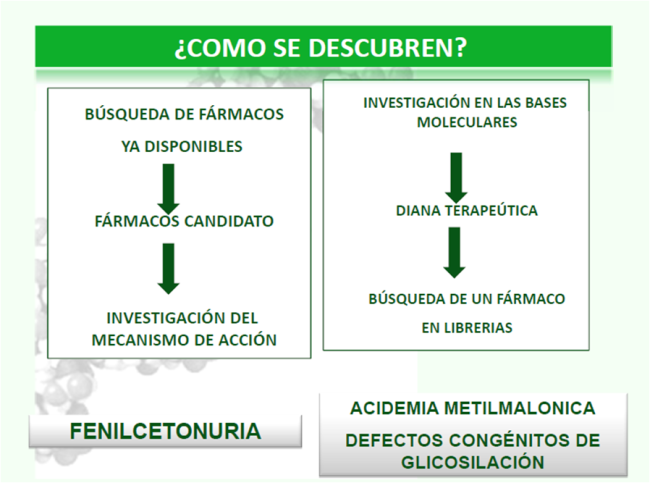
En primer lugar se buscan fármacos ya disponibles, como en el caso de la PKU, en donde existe un fármaco candidato que es el cofactor de la reacción de la fenilalanina hidroxilasa, la tetrahidrobiopterina (BH4), investigándose su mecanismo de acción.
En otros casos, se investigan las bases moleculares de la enfermedad, y cuando se tiene la diana terapéutica, se busca un fármaco adecuado en las librerías de fármacos. Las librerías son repositorios de miles de moléculas que están comercialmente disponibles como potenciales agente terapéuticos.
Ejemplos de este tipo de investigación son las que se están realizando en la acidemia metilmalónica y en los defectos congénitos de la glicosilación.
¿En el caso de la PKU, todos los pacientes responden al tratamiento farmacológico con el cofactor BH4?
Un 30% de pacientes PKU con determinadas mutaciones responden a la administración oral de BH4 (KUVAN®).
En general, son pacientes con PKU leve o moderada, aunque en algunos casos la respuesta es parcial y la terapia con BH4 se debe combinar con la restricción de fenilalanina de la dieta.
¿Podemos predecir en base al genotipo (mutaciones) qué pacientes van a responder a esta terapia?

Se creía inicialmente que las mutaciones que respondían a BH4 eran las que estaban localizadas en el centro activo y que afectaban a la unión de BH4 con la proteína PAH, pero se ha visto que están en todos los dominios (catalítico, regulador y de tetramerización) de PAH y la mayoría están fuera del centro activo y no afectan a la unión de BH4 con la proteína.
No obstante, la respuesta a BH4 depende del genotipo y del estado metabólico del paciente (en estado catabólico la respuesta es menor).Como no todos los pacientes responden a BH4, se están investigando otras chaperonas farmacológicas para PKU.
¿Cuál es el proceso de desarrollo de una chaperona?

En primer lugar se prueba la terapia en células en cultivo del paciente y/o en modelos celulares de la enfermedad. Se prueba a continuación en animales de experimentación para verificar su eficiencia, ausencia de toxicidad y efectos secundarios.
Se realizan ensayos clínicos, que consisten en probar en un pequeño número de pacientes voluntarios (fase I) y en un número mucho mayor (fases II y III).
¿Existen muchas chaperonas farmacológicas?
Existen colecciones de fármacos que cumplen con estas condiciones.
- 10.000 compuestos de alta pureza.
- Librería de compuestos con potencial actividad de chaperona química o farmacológica.
- Son compuestos pequeños.
- Seleccionados por criterios de química médica.
- Sillares químicos susceptibles de modificaciones para desarrollar fármacos.
¿Cómo se realiza la selección de posibles chaperonas farmacológicas?
Como ejemplo, conocemos el estudio de identificación de chaperonas farmacológicas como agentes terapéuticos potenciales para tratar la PKU, realizado por el Dr. Angel Pey con el grupo de la Dra. Aurora Martínez (Pey A et al, J Clin Invest 2008; 118:2858).
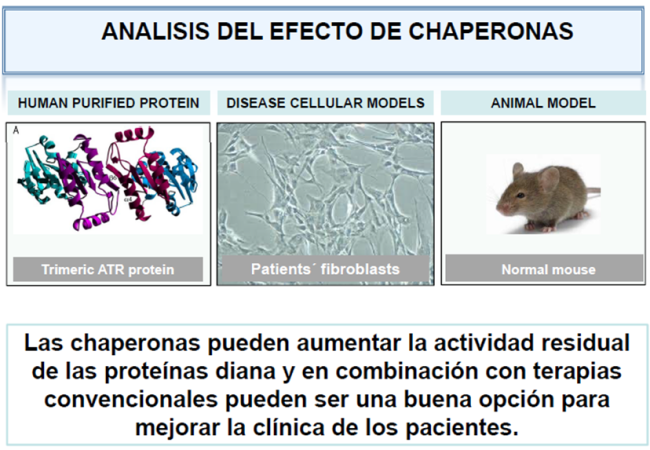
- Se debe disponer de la proteína enzimática fenilalanina hidroxilasa (PAH) purificada.
- Se seleccionan 1000 compuestos potencialmente activos de la librería.
- Se investiga su acción sobre la proteína PAH purificada, seleccionando, por ejemplo, 4 compuestos que:
- Aumentan la estabilidad de la proteína al calor.
- No inhiben su actividad enzimática en unas concentraciones bajas.
- Se seleccionan 2 de los 4 compuestos que además estabilizan la estructura de tetrámero (4 subunidades iguales) de la PAH.
- Se administran pequeñas concentraciones de estos compuestos 12 días a ratones normales para ver si se eleva la actividad PAH en hígado (en este caso no se puede investigar en fibroblastos porque la PAH solo se expresa en hígado).
- Se selecciona el compuesto más activo.
- Se investiga en hígado de ratón mutante, que porta una mutación de plegamiento cuyas mutaciones missense causen defectos de plegamiento a la PAH.
Conclusiones
- Las chaperonas farmacológicas representan una opción terapéutica en las enfermedades metabólicas ya que presentan una buena biodistribución, atraviesan la barrera hematoencefálica y son fácilmente administrables oralmente.
- Previsiblemente son muchos los pacientes que se pueden beneficiar de esta terapia. Ahora bien, sólo los pacientes con mutaciones que alteran conformacionalmente las enzimas son subsidiarios de este tratamiento.
- Las chaperonas farmacológicas puede combinarse con otros tratamientos ya implementados mejorando el pronóstico de los pacientes.
- Los estudios con chaperonas avanzan rápido en la actualidad, sobre todo para aquéllos ECM en los que predominan entre los pacientes aquellas mutaciones que alteran la configuración de la proteína.

Dra. Belén Pérez González. Centro de Diagnóstico de Enfermedades Moleculares. Universidad Autónoma de Madrid.
- 15964 lecturas
También te puede interesar

")