Enfermedades mitocondriales: aspectos actuales de la presentación clínica neonatal y el diagnóstico
Resumen (simplificado) del seminario “NeuroConCiencia” (29/9/2020), Dr. Rafael Artuch, y de “Metabolic Moments” (2/10/2020), Dra. Àngels García-Cazorla, Hospital Sant Joan de Déu Barcelona.

Dentro del campo de las enfermedades metabólicas de base genética, un grupo de trastornos que destaca por su complejidad, tanto en el diagnóstico como en el tratamiento, es de las enfermedades mitocondriales. Son enfermedades raras que en su conjunto afectan al menos a 1 de cada 5.000 individuos, pero es más que probable que sean más frecuentes, pero infradiagnosticadas.
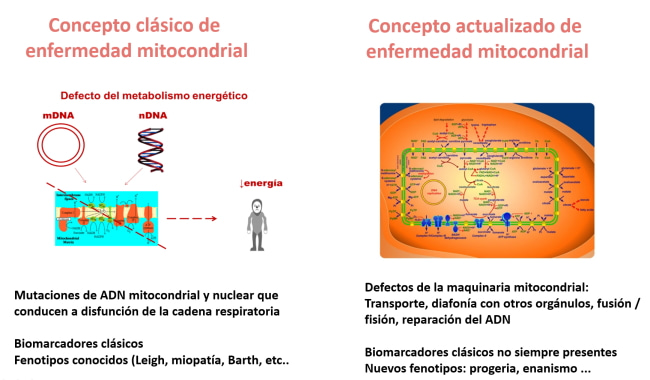
En la actualidad, se han identificado aproximadamente 400 genes cuyas mutaciones son causantes de enfermedades mitocondriales. Si bien es difícil encontrar un punto común para todas estas enfermedades, la falta de energía es probablemente el mecanismo más importante causante de la enfermedad.
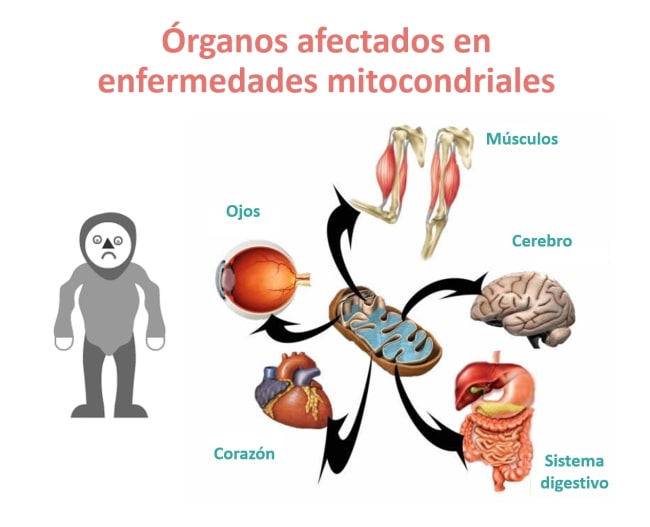
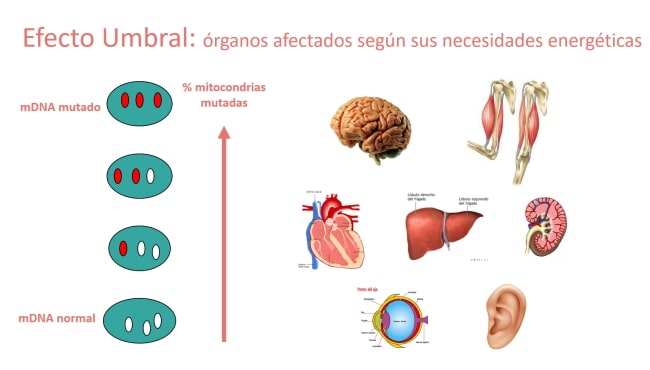
Las mitocondrias son como la cocina de nuestras células y órganos, y cuando se producen fallos, la consecuencia inmediata es la falta de energía, que afectará primordialmente a los órganos más dependientes de ella: músculo, sistema nervioso, corazón, hígado, y a órganos sensoriales como el ojo y el oído. No obstante, cualquier tejido se puede ver implicado en estas enfermedades, dificultando el diagnóstico.
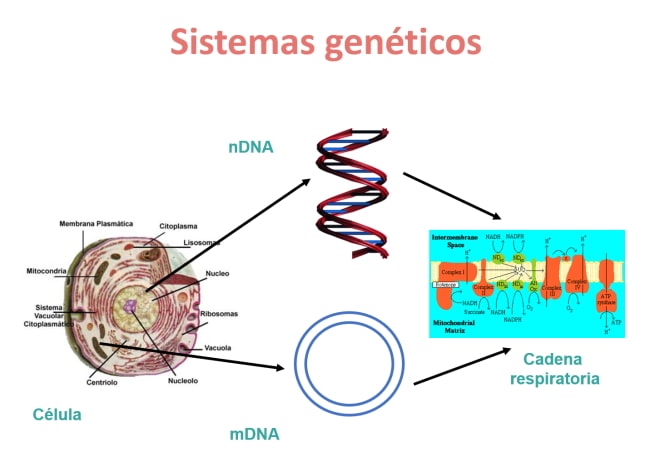
El aspecto más peculiar de las enfermedades mitocondriales es que son las únicas causadas por mutaciones en dos sistemas genéticos. El nuclear, que es el causante de la mayoría de las enfermedades raras, y el mitocondrial. La conjunción de ambos sistemas genéticos es esencial para que funcione la extracción de energía que nuestras células consiguen de los alimentos, y que nos permite crecer, desarrollar y madurar nuestros órganos y cumplir otras funciones biológicas esenciales.
El ADN nuclear contiene miles de millones de lecturas (pares de bases), mientras que el ADN mitocondrial cuenta con 15.000, lo que ejemplifica la diferencia entre ambos genomas. No obstante, a pesar de su pequeño tamaño, el ADN mitocondrial es esencial para la obtención de energía.
Sus características más destacables son:
1. Las mutaciones son de herencia materna (o esporádicas “de novo”) a diferencia de las nucleares que siguen el patrón de herencia mendeliana.
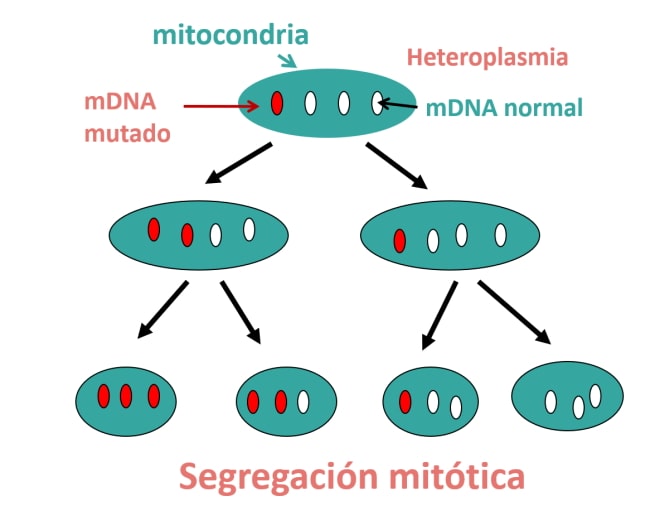
2. Se da el fenómeno llamado heteroplasmia, que significa que las mutaciones del ADN mitocondrial se van a distribuir al azar entre los tejidos y van a tener diferentes proporciones de mutación, desde células prácticamente sanas (con un porcentaje de mutación inferior al 10 %) a células que van a manifestar la enfermedad con porcentajes de mutación que pueden llegar incluso al 100 %.
3. Efecto umbral. Los tejidos más dependientes de energía manifestarán antes la enfermedad con porcentajes de mutación más bajos que tejidos no dependientes.
Síndromes mitocondriales de presentación neonatal
Además de la presentación neonatal clásica (acidosis láctica congénita, síndromes de Leigh, Alpers, Barth, Pearson, etc.), causada por una disfunción de la fosforilación oxidativa (OXPHOS), durante los últimos años se han descrito más de 250 nuevos defectos que afectan a la maquinaria mitocondrial: defectos de transporte, de la interacción de la mitocondria con otros orgánulos, fusión/fisión, reparación del ADN, etc. Además, se han descrito nuevos fenotipos en los que los clásicos biomarcadores no están presentes.
La correlación fenotipo-genotipo es mala en las enfermedades mitocondriales, incluso en síndromes bien definidos. Se han descrito muchos genes mutados nuevos en síndromes neonatales ya conocidos.
Por ejemplo, el síndrome de Leigh (trastorno neurodegenerativo progresivo en la infancia), muestra una marcada variabilidad genética y se asocia con variantes patogénicas en más de 75 genes del ADN mitocondrial o nuclear diferentes. Por el contrario, un solo genotipo puede presentarse con una variedad de fenotipos.
Además, han descrito nuevos fenotipos (enanismo, progeria, etc..) asociados a defectos mitocondriales.
¿Cómo se diagnostican actualmente las enfermedades mitocondriales?
El diagnóstico es difícil por la ausencia de biomarcadores específicos. Los aumentos de lactato, piruvato y alanina (biomarcadores clásicos) son alteraciones orientativas, pero inespecíficas.
Los estudios histológicos y enzimáticos requieren biopsias de los órganos afectados y aun cuando orientan a la existencia y localización de un defecto, no proporcionan el diagnóstico definitivo del mismo.
Teniendo en cuenta que el origen genético de estas enfermedades implica a unos 400 genes patogénicos, las técnicas de secuenciación de nueva generación del exoma y, últimamente, de todo el genoma son las más utilizadas y efectivas. No obstante, la complejidad de estas enfermedades es tal que, aun así, solo se consigue el diagnóstico definitivo en un 50% de los pacientes.
Referencia
Más información en Guía Metabólica
- 5015 lecturas
También te puede interesar


