El trasplante de médula ósea como opción terapéutica en los errores congénitos del metabolismo
")
El trasplante de médula ósea es una opción terapéutica que se ha usado con éxito en Leucodistrofia metacromática, Adrenoleucodistrofia ligada al cromosoma X, Mucopolisacaridosis y en la enfermedad de Krabbe, consiguiendo mejoras en algunos de los trastornos que se manifiestan a causa de estas enfermedades, aunque no es un procedimiento curativo.
De la mano de la Dra. Isabel Badell, del Hospital Sant Joan de Déu y el Hospital Santa Creu i Sant Pau, analizamos este procedimiento y sus aplicaciones.
¿Qué es la médula ósea?
![Médula roja. Imagen: HSJDBCN con foto de Stevenfruitsmaak (Own work) [Public domain], via Wikimedia Commons](/sites/default/files/2015/medula_roja.png) La médula ósea es un tejido que se encuentra en el interior de los huesos largos, vértebras, costillas, esternón, huesos del cráneo, cintura escapular y pelvis.
La médula ósea es un tejido que se encuentra en el interior de los huesos largos, vértebras, costillas, esternón, huesos del cráneo, cintura escapular y pelvis.
La médula ósea puede ser roja y amarilla, según que se aloje en los huesos planos o en los largos, respectivamente.
La médula ósea roja es un tejido esponjoso que se halla en el interior de los huesos planos:
- El esternón
- Las vértebras
- La pelvis y las costillas
Tiene función hematopoyética, es decir, de producción de células sanguíneas.
¿Qué significa hematopoyesis?
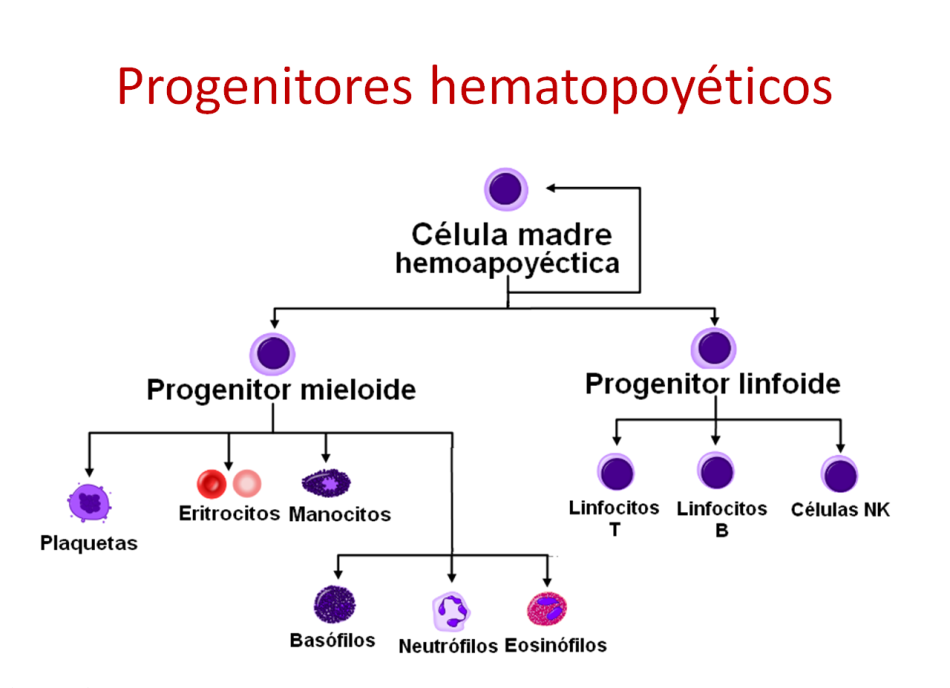
La hematopoyesis es el proceso de formación, desarrollo y maduración de las células de la sangre (eritrocitos, leucocitos y plaquetas) a partir de un precursor celular común e indiferenciado llamado célula madre hematopoyética.
Las células hematopoyéticas tienen la capacidad de implantarse en la médula ósea de un paciente y dar lugar a un sistema inmune sano.
Las células hematopoyéticas se diferencian en tres categorías:
- Células madre, con mayor capacidad de renovación y diferenciación en diferentes células posteriormente.
- Progenitores hematopoyéticos, que tienen restringida su capacidad de diferenciación a otras células, aunque sí pueden renovar un determinado tipo de células.
- Células hematopoyéticas maduras, que son la mayoría de las células presentes en los órganos hematopoyéticos (médula ósea por ejemplo) y constituyen el estadio final de maduración.
¿Qué es el trasplante de médula ósea?
Es un procedimiento que consiste en la extracción de la médula ósea (progenitores hematopoyéticos) de un donante vivo y su transfusión al sistema circulatorio de un receptor.
El trasplante de progenitores hematopoyéticos se emplea para tratar enfermedades que afectan directamente a las células de la sangre (leucemias, inmunodeficiencias,…), para reponer la médula ósea dañada por determinados tratamientos (tumorales o inmunológicos) y en determinadas enfermedades metabólicas lisosomales y peroxisomales.
¿Cuáles son las fuentes de progenitores hematopoyéticos?
La fuente clásica para la obtención de progenitores hematopoyéticos es la médula ósea, pero también se emplean la sangre periférica, la sangre de cordón umbilical y la de hígado fetal. Por ello se acostumbra a hablar del trasplante de médula ósea (TMO), aunque su nombre más correcto sería trasplante de progenitores hematopoyéticos (TPH).
¿Cuáles son los tipos de donantes?
Según el tipo de donante, podemos distinguir el:
- Trasplante autólogo, en el que la fuente de progenitores hematopoyéticos es el mismo paciente.
- Trasplante alogénico, en el que la fuente es una persona diferente, familiar (relacionada) o no familiar.
¿Qué son los antígenos leucocitarios humanos (HLA)?
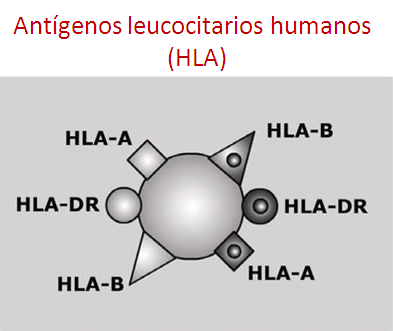 Los antígenos leucocitarios humanos (HLA) son moléculas localizadas en la superficie de las células y determinan el reconocimiento inmunológico.
Los antígenos leucocitarios humanos (HLA) son moléculas localizadas en la superficie de las células y determinan el reconocimiento inmunológico.
Es decir, el reconocimiento de nuestras propias células como nuestras y las células ajenas como “no nuestras” o extrañas.
Están implicadas en la tolerancia y rechazo al TPH.
La compatibilidad HLA completa requiere que donante y receptor presenten los mismos antígenos a nivel proteico (serológico) y a nivel genético, en los dos alelos que codifican a HLA (alélico).
Es decir, que el donante y el receptor tengan las mismas moléculas HLA en la superficie de las células, y también la misma información en su DNA para la formación de nuevas moléculas de HLA de superficie.
Tipos de TPH según la compatibilidad del donante
- Trasplante singénico es aquel en que el donante es un hermano gemelo univitelino (igual información genética para HLA).
- Trasplante alogénico de familiar HLA idéntico es aquel en que el familiar es totalmente compatible.
- Trasplante alogénico de familiar HLA no idéntico, cuando existen diferencias en el sistema HLA.
- Trasplante no familiar HLA idéntico.
- Trasplante no familiar HLA no idéntico.
Cuanto mayor es la compatibilidad del donante, mayor es la probabilidad de que no se produzca un rechazo y menor la posibilidad de presentarse la enfermedad del injerto contra el receptor.
¿Qué es la enfermedad del injerto contra el receptor o huésped (EICH)?
La EICH se produce cuando el sistema inmunológico del donante reconoce los antígenos del receptor como extraños y genera una reacción frente a él. ircibegre delades geno familiar. y genera una rracciSe estima que la EICH aparece en el 30-50% de los TPH familiares HLA idénticos y en el 60% de los TPH no idénticos. El riesgo de EICH limita el uso de un donante no familiar, en enfermedades genéticas.
Extracción de médula ósea y obtención de progenitores hematopoyéticos
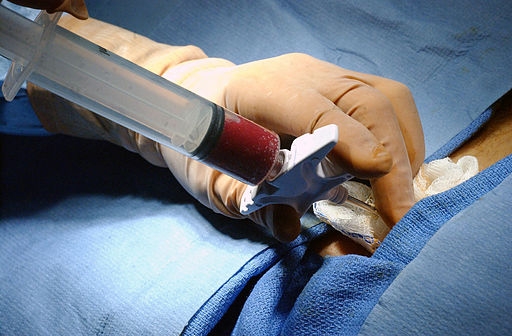 La extracción de la médula osea se realiza mediante una punción aspirativa en las crestas ilíacas (borde superior del hueso ilíaco donde hay mayor cantidad de médula ósea), extrayéndose entre 800-1200 ml de sangre en el adulto y 10-20 ml/kg de peso en el niño. En la médula ósea hay 1-2% de células madre y progenitores hematopoyéticos.
La extracción de la médula osea se realiza mediante una punción aspirativa en las crestas ilíacas (borde superior del hueso ilíaco donde hay mayor cantidad de médula ósea), extrayéndose entre 800-1200 ml de sangre en el adulto y 10-20 ml/kg de peso en el niño. En la médula ósea hay 1-2% de células madre y progenitores hematopoyéticos.
Para obtener los progenitores hematopoyéticos existen dos posibilidades: a partir de sangre periférica o de sangre del cordón umbilical.
En sangre periférica (es la sangre que se extrae con una punción normal, por ejemplo para una analítica), la proporción de progenitores hematopoyéticos es muy baja (0,01-0,1%), por lo que conviene aumentar dicha proporción.
Para ello el donante es tratado con factores estimulantes, que hacen migrar (salir) a los progenitores hematopoyéticos desde la médula ósea a la sangre periférica, siendo recogidos y seleccionados mediante aféresis (separación y selección de los componentes sanguíneos necesarios).
La sangre del cordón umbilical de los recién nacidos contiene progenitores hematopoyéticos inmaduros y capaces de repoblar los diferentes linajes de células hematopoyéticas. Por ello, actualmente es una buena fuente de células para el trasplante en adultos y especialmente en niños.
¿Se debe acondicionar (preparar) al receptor del TPH?
Previamente al TPH, se debe preparar la médula ósea del paciente para la recepción del nuevo sistema hematopoyético del donante, creando un espacio medular. Se puede realizar mediante irradiación o quimioterapia del paciente que va a recibir el trasplante.
¿Tiene limitaciones el TPH?
Las limitaciones fundamentales son la toxicidad del acondicionamiento, la dificultad de hallar el donante adecuado en un tiempo reducido y la imposibilidad de alcanzar a ciertos tejidos no hematopoyéticos, como el hueso o el sistema nervioso.
El TPH en los ECM
El TPH como opción terapéutica en los ECM se basa en el fenómeno de corrección cruzada, el origen hematopoyético de la microglia (células gliales que actúan como macrófagos en cerebro y la médula ósea con función de defensa inmune) y la evidencia de que una pequeña cantidad de enzima activa puede corregir una deficiencia enzimática lisosomal. Es decir, en el sistema nervioso hay células que actúan como defensas y que podrían beneficiarse de esta terapia, ya que su origen es hematopoyético.
¿Qué es el fenómeno de corrección cruzada?
Consiste en la corrección de los defectos enzimáticos lisosomales “in vitro” al cultivar conjuntamente fibroblastos de pacientes con enfermedades de Hurler y Hunter (descubierta por Fratantoni y col. en 1969). La transferencia cruzada fuera del tejido hematopoyético determina la mejoría de los síntomas clínicos, muy evidente en los órganos reticuloendoteliales (hígado y bazo).
¿Qué papel tiene la microglía?
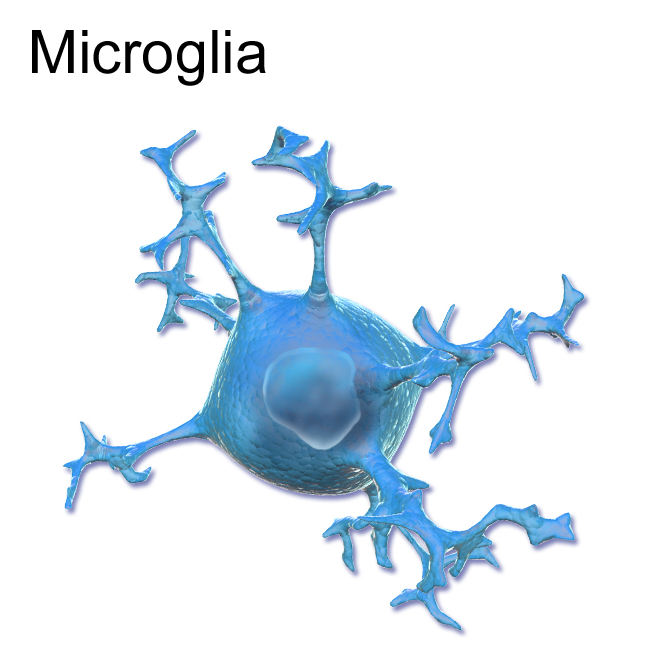 Existe evidencia que las células monocíticas (un tipo de células sanguíneas) del paciente receptor pueden atravesar la barrera hematoencefálica (que separa y defiende el sistema nervioso de la sangre) y transformarse en células microgliales.
Existe evidencia que las células monocíticas (un tipo de células sanguíneas) del paciente receptor pueden atravesar la barrera hematoencefálica (que separa y defiende el sistema nervioso de la sangre) y transformarse en células microgliales.
La producción de microglía que procede de los monocitos del donante es una fuente constante de la enzima deficiente del receptor y permite la utilización de ésta por sus neuronas.
No obstante, el recambio de la microglia es más lento que el que ocurre en los órganos retículoendoteliales (hígado y bazo), por lo que el control de los síntomas del sistema nervioso es lento y tardío.
En algunas enfermedades parece que se requiere al menos de 6 meses a un año para observar mejoría en las alteraciones neurológicas.
El tratamiento en estos casos debería ser precoz, para evitar el deterioro neurológico durante el primer año post-trasplante.
Factores que influyen en el pronóstico
Los principales factores son el tipo de enfermedad (no todas las enfermedades lisosomales responden igual al TPH), genotipo (mutaciones más graves tienen menor respuesta), edad (a menor edad, mejor resultado), eficacia y complicaciones del propio trasplante.
El TPH no es curativo, si no que mejora los trastornos sistémicos de la enfermedad. Por ello el paciente trasplantado deberá seguir un control médico multidisciplinario para evitar o prevenir las complicaciones.
¿Cuáles son las fuentes de progenitores hematopoyéticos más usadas en el TPH en ECM?
Aunque las diferentes fuentes (médula ósea, sangre de cordón y sangre periférica) no han mostrado diferencias en los porcentajes de injerto, tradicionalmente se ha usado el TMO, a partir de un donante idéntico familiar.
Sin embargo, la sangre de cordón consigue altas tasas de quimera completa (hematopoyesis del donante por encima del 90%). Es preferible que el donante no sea portador de la deficiencia enzimática para alcanzar mejor actividad enzimática en el receptor.
TPH en enfermedades lisosomales
Mucopolisacaridosis
Las mucopolisacaridosis son enfermedades multisistémicas causadas por mutaciones en genes que codifican a enzimas lisosomales que degradan mucopolisacáridos o glucosaminoglicanos.
El TPH se ha utilizado con buenos resultados en muchos pacientes con MPS, especialmente MPS I antes de los dos años. Mejora el aspecto físico (la dismorfia facial y el crecimiento), la opacidad corneal se estabiliza, mejora la audición y la hepatoesplenomegalia. No obstante, las alteraciones esqueléticas (MPS IV) y oculares persisten y no parece mejorar la función intelectual.
El TPH se ha realizado en más de 500 pacientes con MPS I (Hurler). Se puede aplicar combinado con la terapia enzimática sustitutiva (enzima sintetizada por biotecnología, tanto en MPS I, como en MPS II y MPS VI.
Enfermedad de Krabbe
La enfermedad de Krabbe está causada por la deficiencia de la enzima lisosomal galactocerebrosidasa. Este defecto causa la acumulación anormal de un esfingolípido no degradado, el galactocerebrósido, que da lugar a células de aspecto globoide en el sistema nervioso central y periférico.
El TPH se puede aplicar en la forma precoz durante el primer mes de vida, aunque persisten los problemas motores. En las formas tardías, aplicado al inicio de los síntomas, estabiliza las manifestaciones neurológicas.
Leucodistrofia metacromática
La leucodistrofia metacromática es una enfermedad metabólica debida a la deficiencia de la enzima lisosomal arilsulfatasa A. El TPH se aplica en formas tardías presintomáticas, con buena función neuropsicológica.
TPH en enfermedades peroxisomales
Adrenoleucodistrofia ligada al cromosoma X
La adrenoleucodistrofia ligada al cromosoma X es una enfermedad peroxisomal causada por mutaciones en el gen ABCD1 que codifica la proteína ALDP, que participa en la β–oxidación de los ácidos grasos de cadena muy larga.
El TPH se puede aplicar en la forma cerebral inicial pudiendo estabilizar la función neurológica y neurocognitiva.
Más información sobre TPH:
- González Gutiérrez-Solana L, Pérez Martínez A, Cantarín Extremera V. Trasplante de progenitores hematopoyéticos en los errores congénitos del metabolismo. En: Sanjurjo P, Baldellou A (eds) Diagnóstico y tratamiento de las enfermedades metabólicas hereditarias. Ergon, 4ª edición, 2014. Cap 17, pp: 279-298.
- Aldenhoven M, Kurtzberg J. Cord blood is the optimal graft source for the treatment of pediatric patients with lysosomal storage diseases: clinical outcomes and future directions. Cytotherapy 2015 Mar 31. pii: S1465-3249(15)00693-3.
- Boelens JJ, Orchard PJ, Wynn RF. Transplantation in inborn errors of metabolism: current considerations and future perspectives. Br J Haematol 2014;167(3):293-303.
Equipo Guía metabólica. Revisado por la Dra. Isabel Badell, Hospital Sant Joan de Déu y Unidad Pediátrica de TPH Hospital Santa Creu i Sant Pau.
Imágenes:
Comprobando muestra de sangre. National Eye Institute en Flickr (CC BY-NC-ND 2.0)
Antígenos leucocitarios humanos (HLA). Imagen: Fernando da Rosa en Wikimedia
Biopsia de médula osea: Imagen: Photographer's Mate 2nd Class Chad McNeeley. Navy News Service via Wikimedia Commons
Microglia. Imagen: Blausen.com staff. "Blausen gallery 2014". Wikiversity Journal of Medicine. DOI:10.15347/wjm/2014.010. ISSN 20018762.
- 8799 lecturas
También te puede interesar


