Opciones terapéuticas actuales: I /V (04-2011)

Con este capítulo comenzamos una serie de documentos orientados a explicar de forma sencilla el mecanismo de acción de las opciones terapéuticas más recientes, en algunos casos, tratamientos prometedores para ciertos errores congénitos de metabolismo.
En concreto hablaremos de chaperonas farmacológicas, tratamiento enzimático sustitutivo, inhibidores de sustrato, terapia génica y trasplante.
Introducción. Estructura básica de las proteínas.
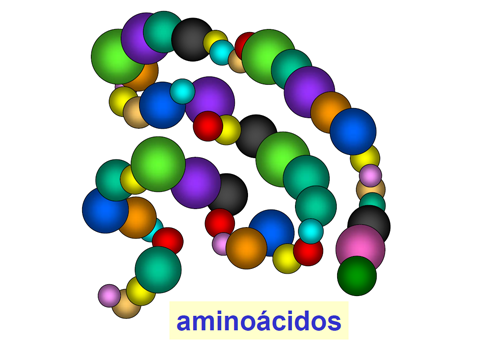
Las proteínas son largas cadenas de aminoácidos unidos entre sí. Hay 20 aminoácidos comunes que forman todas las proteínas que se presentan en la naturaleza. Estos 20 aminoácidos son diferentes entre sí por sus características bioquímicas.
Para que esta cadena de aminoácidos pueda desarrollar su función como proteína ha de plegarse de forma más o menos compleja en una estructura tridimensional.
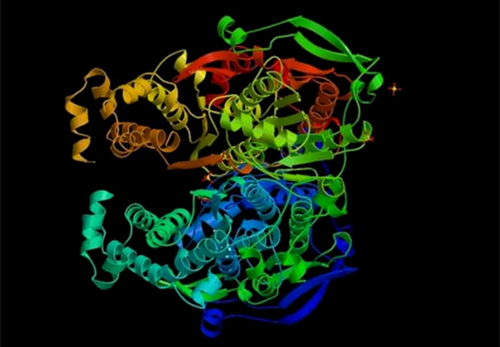
Esta configuración “esconderá” aquellas zonas más vulnerables y permitirá que las zonas activas (los centros catalíticos), se sitúen y formen de manera adecuada.
Si se llega a obtener un buen plegamiento y una buena estructura tridimensional, la estabilidad de la proteína será la adecuada, así como su función.
El centro catalítico es la zona de la proteína con la actividad funcional específica, el resto de la proteína tiene una función de estructura.
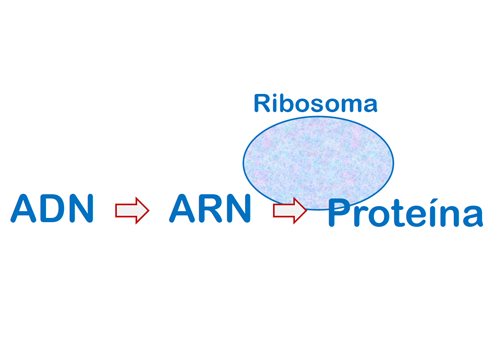
Las proteínas se forman a partir de la información que reside en los genes, en el ADN que se encuentra en el núcleo celular. Los ribosomas son pequeños orgánulos encargados de ir formando la cadena de aminoácidos, que luego será una proteína, a partir de la información que llega del ADN en forma de ARN.
La cadena de aminoácidos debe pasar por ciertas transformaciones antes de ser una proteína funcional, como el plegamiento o la glicosilación (unión de azúcares a la proteína).
Mecanismos de control de calidad de las proteínas a nivel celular
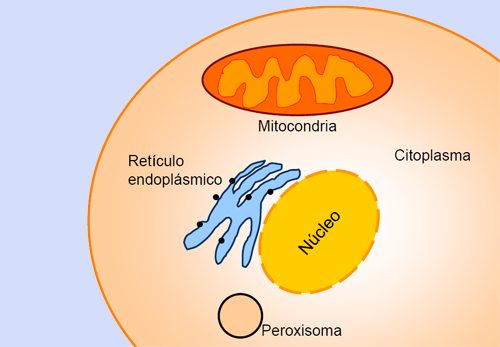
Desde que la proteína se forma como cadena de aminoácidos hasta que adquiere su estructura final y llega al compartimento celular en el que actúa ha de ser protegida.
Para evitar esto las células poseen un complejo sistema de protección de proteínas. Este sistema de protección está formado por proteínas de control que se dedican a ayudar al adecuado replegado tridimensional así como al transporte de las proteínas emergentes o recién formadas.
Hay proteínas de control que aumentan ante un estrés a nivel celular y que actúan como acompañantes o “chaperonas”. Toman las proteínas dañadas, las repliegan de nuevo de forma correcta y derivan aquéllas proteínas demasiado dañadas a los sistemas de degradación celular.
Este llamado sistema de control de calidad está formado por gran cantidad de proteínas, no sólo chaperonas, también hay chaperoninas, cochaperonas, chaperonas-lectinas, proteasas… algunas de estas proteínas se localizan en el retículo endoplásmico, otras en el citosol, otras en la mitocondria e incluso algunas en los peroxisomas.
Cuanto más compleja es una proteína más necesidad de ayuda por parte de las chaperonas necesita. Cuando hay mutaciones o proteínas anómalas afectándose la forma de plegarse la proteína, el sistema de control de calidad puede ayudar a un adecuado plegado.
Enfermedades y alteraciones en las proteínas
Según la alteración en la estructura proteica se han descrito diferentes enfermedades llamadas “enfermedades conformacionales” (conformational diseases):
- Enfermedades en las que lo que predomina es una degradación muy aumentada de proteínas mal plegadas. A este grupo pertenecerían aquéllos errores congénitos del metabolismo que se deben a alteraciones estructurales de las proteínas o “conformacionales”.
Otras menos frecuentes en errores congénitos del metabolismo:
- Enfermedades en las que las proteínas mal plegadas no se agregan pero ejercen una dominancia negativa, es decir, influyen de forma negativa en la función de las proteínas “iguales” pero normales. Esto pasa en enfermedades cardíacas hereditarias y en enfermedades de la queratina. La queratina alterada ejerce un efecto negativo frente a la queratina que se forma normalmente.
- Enfermedades en las que las proteínas anómalas o mal plegadas tienden a agregarse, es el caso de la enfermedad de Alzheimer y otras.
- Por último, hay enfermedades que se deben a problemas en el propio sistema de control de calidad de las proteínas. En estas enfermedades pueden coincidir alteraciones en diferentes proteínas porque no son adecuadamente “ayudadas” por las chaperonas y similares. Algunos casos de Enfermedad de Leigh, otras enfermedades mitocondriales y enfermedades peroxisomales pueden pertenecer a este último grupo.
Errores congénitos del metabolismo y plegado anormal de las proteínas

Los errores congénitos del metabolismo se deben a mutaciones (cambios) en un gen que da lugar a una proteína anómala o defectuosa.
Los genes son cadenas de bases nitrogenadas que forman una doble hélice.
Un gen puede tener miles de bases nitrogenadas.
El 90% de las mutaciones se deben a pequeños cambios como mutaciones puntuales (cambio de una base por otra equivocada. Mirando al esquema de al lado, una base de un color, por otro), pequeñas inserciones de bases (aparecen una o varias nuevas bases donde no había) o pequeñas deleciones (desaparecen alguna o varias bases nitrogenadas).
Las mutaciones puntuales pueden producir cambios en la proteína que podemos dividir en dos grupos:
- Mutaciones que dan lugar a cambio de un solo aminoácido de la cadena. Estas mutaciones pueden alterar el plegamiento proteico y alterar la función.
- Mutaciones que producen una cadena de aminoácidos más corta de lo normal, o cambian toda la cadena de aminoácidos a partir de un punto…
En ambos casos el resultado funcional depende de la actividad de la propia proteína mal formada y de la capacidad del sistema de control de calidad (chaperonas) para evitar que sea degradada y plegarla de forma adecuada.
A priori podríamos pensar que la propia mutación genética determina que el paciente que la padece tenga más o menos manifestaciones clínicas, sin embargo, esto en realidad no siempre es así. Dos pacientes con la misma mutación pueden ser bien diferentes.
Probablemente depende, entre otros factores, de que el sistema de control de calidad funcione de forma correcta. Para una mutación concreta las manifestaciones de un paciente serán leves si funciona bien su sistema de control de calidad, mientras que para otro pueden ser muy severas si su sistema de control de calidad es defectuoso.
Opciones terápeuticas
Puesto que los defectos en el plegado proteico parecen la base en algunos casos de errores congénitos del metabolismo, las llamadas proteínas chaperonas pueden resultar de utilidad en el tratamiento de los mismos. Estas proteínas se administran de forma exógena (externa), protegen las proteínas anómalas de ser degradadas y les ayudan a plegarse de forma optimizada. Se denominan chaperonas farmacológicas.
De momento su uso es básicamente experimental, salvo en casos concretos como la Fenilcetonuria, donde el uso de tetrahidrobiopterina (BH4) tiene un efecto de chaperona farmacológica.
Los estudios en enfermedades lisosomales son muchos, algunos de ellos prometedores. En concreto se ha demostrado un defecto de plegado proteico en enzimas lisosomales propias de la enfermedad de Fabry, Gaucher, gangliosidosis GM1 y GM2, enfermedad de Pompe y mucopolisacaridosis IIIB. Para estas enfermedades hay estudios en la última década con chaperonas farmacológicas. Estos estudios se encuentran en diferentes estadios de desarrollo.
En defectos de la oxidación de los ácidos grasos también se han descrito enfermedades debidas a un defecto de plegado proteico así como también en algunos defectos del metabolismo de la homocisteína.
Pero el uso de “chaperonas” no se restringe a los errores congénitos del metabolismo, de forma que la experiencia en otras enfermedades más comunes, como la fibrosis quística por ejemplo, servirá para avanzar en experiencia y manejo terapéutico con estas sustancias.
Como conclusión vamos a concretar en ejemplos sencillos:

Si una proteína anómala tiene un 5% de la actividad que debería tener y conseguimos, con una chaperona, que se mantenga estable y activa 3 veces más tiempo de lo que sería capaz sola, conseguiremos que alcance 15% de la actividad de una proteína normal, porque prolongamos su vida media.
Si una proteína, por su alteración estructural, tiene más difícil su actividad, la chaperona facilitaría la interacción con el sustrato. Si la proteína fuese la cerradura, el sustrato sería la llave y la chaperona, el “3 en 1”.
Documentos
Dras. Mercedes Serrano y Maria Antònia Vilaseca Unidad de Enfermedades Metabólicas Hospital Sant Joan de Déu
- 2207 lecturas
Noticias relacionadas

También te puede interesar


