Mutaciones en el gen GOT2 causan una encefalopatía relacionada con la lanzadera malato-aspartato
Resumen de la Cápsula metabólica, presentada por la Dra. Àngels García-Cazorla, en la Unidad de Enfermedades Metabólicas Congénitas, Hospital Sant Joan de Déu Barcelona, el 22 de enero de 2021.

El gen GOT2 codifica la isoforma mitocondrial de la enzima glutamato oxalacetato transaminasa (GOT; EC 2.6.1.1), mientras que GOT1 codifica la isoforma citosólica. Ambas isoformas catalizan la interconversión reversible de oxalacetato y glutamato en aspartato y α-cetoglutarato y forman parte de la lanzadera malato-aspartato (LMA).
¿Qué es la lanzadera malato-aspartato?
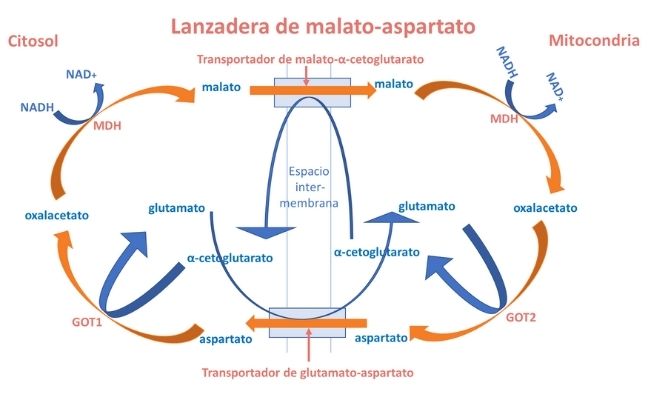
La lanzadera malato-aspartato (LMA) es un complejo formado por dos transportadores de membrana y 4 enzimas (2 unidades de malato deshidrogenasa (MDH) y 2 unidades de glutamato oxalacetato transaminasa (GOT). Tiene un papel clave en la homeostasis redox intracelular de NAD:NADH (NAD+: nicotinamida adenina dinucleótido; NADH: NAD reducida).
¿Cuál es la función de la lanzadera malato-aspartato?
Su función es el transporte de electrones del NADH del citosol a la mitocondria. El NADH es una coenzima de oxido-reducción presente en todas las células vivas.
El NADH producido en reacciones citosólicas de las deshidrogenasas ligada a NAD, principalmente durante la glucólisis, se vuelve a oxidar a NAD+ dentro de las mitocondrias. Dado que la membrana mitocondrial interna es relativamente impermeable a NAD+ y NADH, existen lanzaderas redox capaces de intercambiar electrones y protones (H+).
La lanzadera de malato-aspartato constituye un mecanismo para la transferencia neta de equivalentes reducidos NADH a través de la membrana mitocondrial.
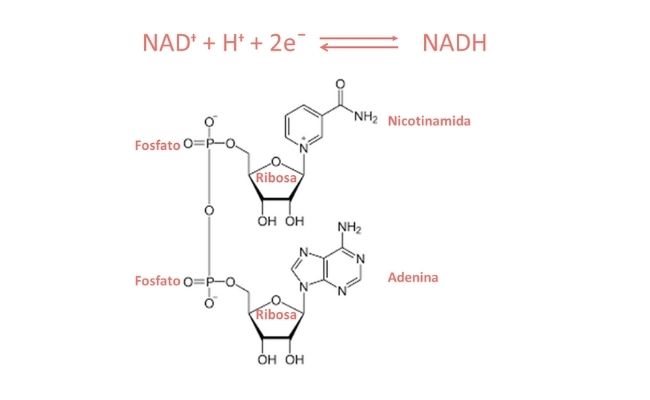
¿Qué ocurre si se produce un defecto en la lanzadera malato-aspartato?
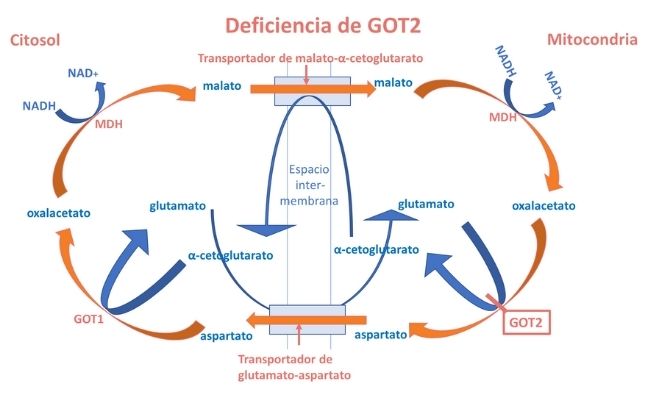
Se altera la transferencia de equivalentes reducidos a través de la membrana mitocondrial y, por tanto, la homeostasis redox intracelular. Mutaciones en los genes que codifican los transportadores y enzimas componentes de la lanzadera dan lugar al mal funcionamiento de la misma.
Se conocen defectos de malato deshidrogenasa (MDH2) y de los dos transportadores (SLC25A12, SLC25A13) y recientemente se han descrito 4 pacientes de 3 familias con mutaciones en GOT2.
¿Qué fenotipo clínico tenían los pacientes con defecto de GOT2?
Los pacientes mostraban microcefalia congénita, bajo peso al nacer, hipotonía neonatal y dificultades para alimentarse. Convulsiones que comienzan entre los 4 y 9 meses: mioclónicas, generalizadas y tónico-clónicas generalizadas, mirada hacia arriba, microcefalia progresiva y resonancia magnética cerebral anormal.
¿Tenían marcadores bioquímicos?
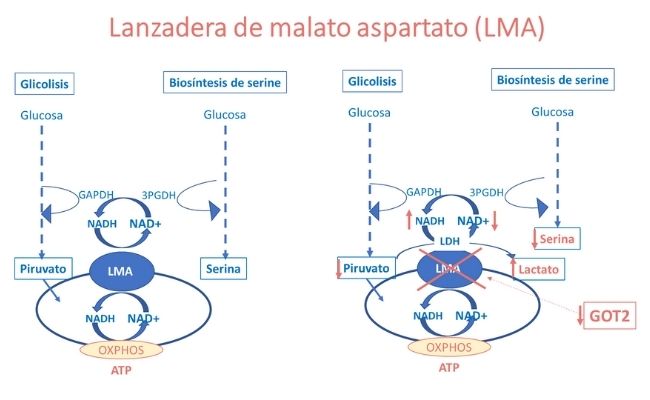
Todos ellos mostraban hiperlactacidemia e hiperamonemia moderadas, pero solo uno de ellos, con clínica más grave, mostraba una baja concentración plasmática de serina y una hipercitrulinemia. Los ácidos orgánicos y acilcarnitinas eran normales en todos ellos.
La hiperlactacidemia y la deficiencia de serina se explican por la alteración de la relación NADH:NAD causada por la deficiencia de GOT2 y con ello, de la función de la lanzadera de malato-aspartato (LMA), tal como de observa en la figura superior.
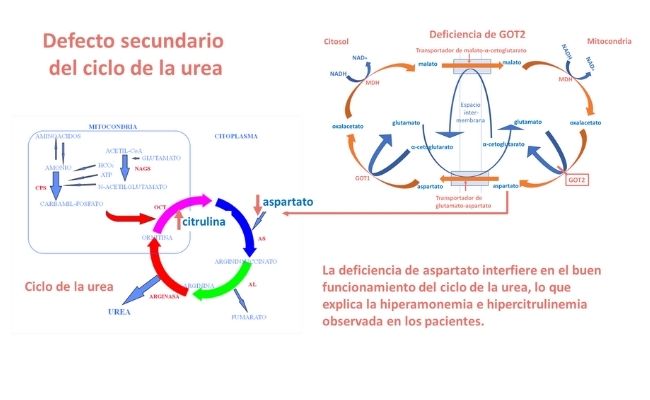
Por otra parte, la deficiente síntesis de aspartato debida al defecto de GOT2 causa una deficiencia de este aminoácido, que es indispensable para la función enzimática de la argininsuccinato sintetasa, enzima del ciclo de la urea, lo que explica la hiperamonemia e hipercitrulinemia moderadas observada en los pacientes, como se observa en la siguiente figura.
¿Cómo se trataron los pacientes?
El paciente más grave solo dejó de tener convulsiones cuando se le administró una combinación de serina y piridoxina. El piridoxal-fosfato es una coenzima indispensable de todas las transaminasas, incluida GOT2. Por ello, puede estimular la actividad residual de GOT2 posiblemente mejorando el plegamiento adecuado de la proteína enzimática GOT2.
Otro paciente fue tratado únicamente con piridoxina durante 8 meses. Su epilepsia estuvo mejor controlada y mejoraron sus funciones cognitivas y su estado de alerta. No obstante, cuando se agregó la suplementación con serina al tratamiento, las convulsiones se controlaron por completo, con una mejor cognición y actividad física. En un período en el que la serina no estuvo disponible, las convulsiones regresaron y desaparecieron nuevamente cuando se reintrodujeron las convulsiones.
La piridoxina es esencial para la síntesis de serina de novo, por lo que los mecanismos de respuesta de la piridoxina pueden deberse en parte a un efecto directo que impulsa la síntesis de serina además de restaurar la homeostasis NADH/NAD+.
Conclusiones
- La deficiencia de GOT2 causa una encefalopatía metabólica con epilepsia de inicio precoz.
- Es una enfermedad metabólica de la lanzadera malato-aspartato, que da lugar a un defecto de la biosíntesis de serina.
- Todos los pacientes presentan hiperlactacidemia e hiperamonemia moderadas.
- El paciente más gravemente afectado mostraba baja concentración plasmática de serina y elevada de citrulina.
- La enfermedad es tratable con suplementación de serina y piridoxina.
- Los modelos animales con defecto total de GOT2 no son viables pero el modelo animal de pez cebra con defecto parcial de GOT2 responde al tratamiento con serina/piridoxina, con supervivencia y control de la epilepsia.
Referencias
- Van Karnebeek CDM, Ramos RJ, Wen XY, Tarailo-Graovac M, Gleeson JG, Skrypnyk C, Brand-Arzamendi K, Karbassi F, Issa MY, van der Lee R, Drögemöller BI, Koster J, Rousseau J, Campeau PM, Wang Y, Cao F, Li M, Ruiter J, Ciapaite J, Kluijtmans LAJ, Willemsen MAAP, Jans JJ, Ross CJ, Wintjes LT, Rodenburg RJ, Huigen MCDG, Jia Z, Waterham HR, Wasserman WW, Wanders RJA, Verhoeven-Duif NM, Zaki MS, Wevers RA. Bi-allelic GOT2 Mutations Cause a Treatable Malate-Aspartate Shuttle-Related Encephalopathy. Am J Hum Genet. 2019 Sep 5;105(3):534-548.
- Borst P. The malate-aspartate shuttle (Borst cycle): How it started and developed into a major metabolic pathway. IUBMB Life. 2020;72(11):2241-2259.
- 13605 lecturas
También te puede interesar


