Hiperamonemia: Actualización del diagnóstico diferencial
Resumen de la Cápsula metabólica presentada por la Dra. Àngels García-Cazorla en la Unidad de Enfermedades Metabólicas del Hospital Sant Joan de Déu Barcelona el 5 de febrero de 2021.

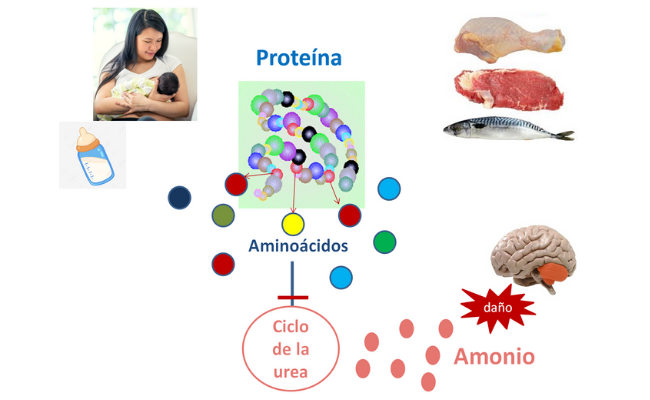
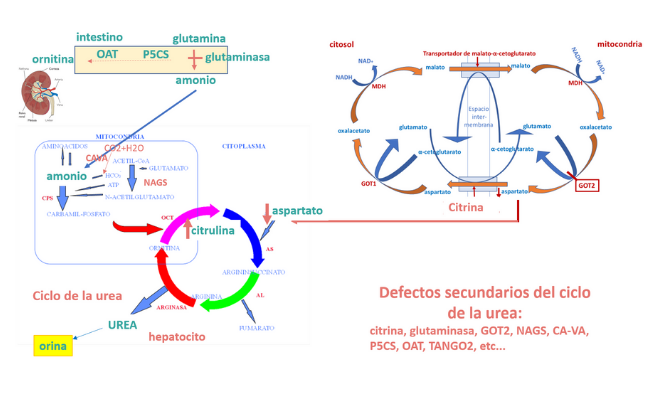
El amonio (NH4+) es un producto neurotóxico del metabolismo de las proteínas y otros compuestos nitrogenados. Mediante el ciclo de la urea, el amonio se transforma en urea para evitar su toxicidad. La urea se elimina por la orina.
¿Qué es la hiperamonemia?
Se considera hiperamonemia la concentración plasmática de amonio superior a 100 µmol/L en neonatos y superior a 50 µmol/L en niños mayores de 1 mes de vida.
¿Por qué es neurotóxica la hiperamonemia?
El amonio se transforma en glutamina en los astrocitos cerebrales, causando edema cerebral. Por ello, la presentación clínica es predominantemente neurológica: inicialmente rechazo de la ingesta y vómitos, posteriormente hipoactividad, letargia y coma. En algunos casos pueden asociarse convulsiones.
Se trata de una patología grave, por lo que su sospecha y detección precoces son esenciales, así como el diagnóstico de su posible causa, pues de ellas depende el pronóstico.
¿Cuáles son las causas de hiperamonemia?
Las causas de hiperamonemia se dividen en:
- Primarias, si se originan en un defecto genético de alguna de las enzimas del ciclo de la urea.
- Secundarias, que pueden ser de origen genético, causadas por otros defectos genéticos que interfieren en el ciclo de la urea, o adquiridas, por inhibición del ciclo de la urea causada por medicación u otras causas clínicas.
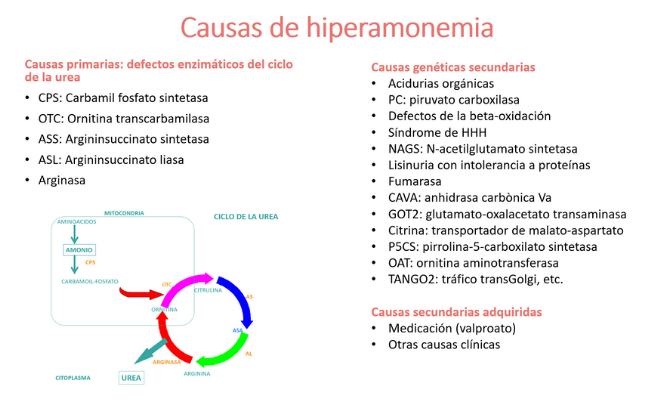
Las causas genéticas primarias y muchas de las secundarias se conocen desde hace años (por ejemplo: acidurias orgánicas, defectos de la beta-oxidación, lisinuria con intolerancia a proteínas, síndrome de HHH), pero algunas de ellas han sido descritas en las últimas décadas (CA-VA, GOT2, citrina, P5CS, TANGO2), de manera que el diagnóstico diferencial se debe ir actualizando periódicamente.
¿Cómo se diagnostica la hiperamonemia?
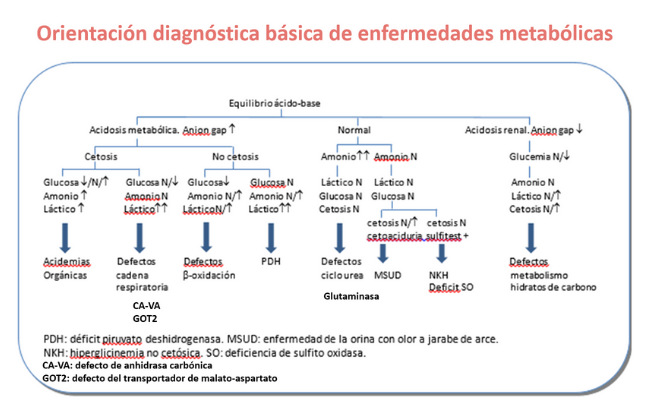
La primera orientación diagnóstica se basa en una serie de parámetros bioquímicos básicos que se realizan (junto con el amonio) en el laboratorio de urgencias (hemograma, equilibrio ácido-base, ionograma, anión gap, glucemia, coagulación, ácido láctico, función hepática y renal, CPK, ácido úrico, cetonemia).
Existe un primer algoritmo orientativo basado en el equilibrio ácido-base y la presencia o ausencia de acidosis metabólica o cetosis.
¿Cómo se alcanza el diagnóstico definitivo?
A esta orientación diagnóstica previa le debe seguir el diagnóstico diferencial. Es importante recoger sangre y orina antes del tratamiento para realizarlo mediante análisis específicos (aminoácidos, ácidos orgánicos, acilcarnitinas, ácidos grasos libres, etc.), en el laboratorio especializado en enfermedades metabólicas.
Es importante también disponer de sangre para preparar DNA, ya que el estudio genético permitirá el diagnóstico definitivo posterior (en un caso neonatal grave o una descompensación grave no diagnosticada anteriormente).
Existen algoritmos específicos de diagnóstico de hiperamonemia, como el basado en los niveles de glutamina (ver referencia: J Inherit Metab Dis. 2019;42: 1192–1230), que deben ser actualizados periódicamente, ya que se van describiendo nuevas enfermedades metabólicas que cursan con hiperamonemia moderada, que deben incluirse en el algoritmo.
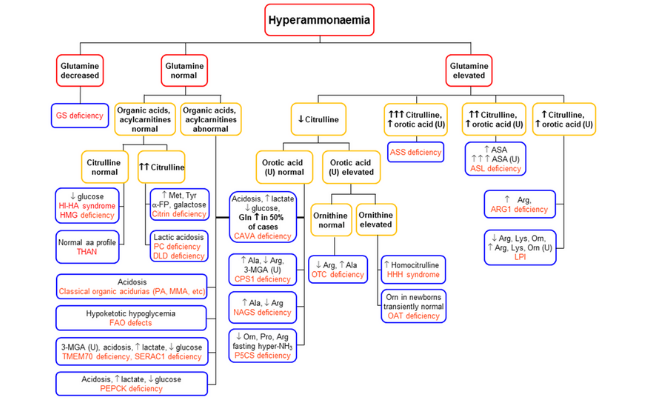
Es indispensable el estudio genético para confirmar el diagnóstico, ya que solo el diagnóstico genético permitirá el consejo genético y el diagnóstico prenatal, si se requiere.
Actualmente el estudio genético se puede realizar mediante paneles génicos, siendo de especial interés en los defectos secundarios en los que el diagnóstico diferencial puede ser más lento y complicado.
Conclusiones
- La hiperamonemia es una patología grave, por lo que su sospecha y detección precoces son esenciales, así como el diagnóstico de su posible causa que permita el tratamiento de emergencia, pues de todo ello depende el pronóstico.
- Las causas principales de hiperamonemia pueden ser primarias, si se originan en un defecto genético de alguna de las enzimas del ciclo de la urea y secundarias, causadas por otros defectos genéticos que interfieren en dicho ciclo metabólico.
- El diagnóstico diferencial se basa en análisis específicos (aminoácidos, ácidos orgánicos, etc). Los algoritmos diagnósticos se deben actualizar periódicamente, ya que se van identificando nuevas enfermedades metabólicas que cursan con hiperamonemia.
- El estudio genético es indispensable para confirmar el diagnóstico y permite el consejo genético y diagnóstico prenatal, si se requiere.
Referencias
- Häberle J, Burlina A, Chakrapani A, et al. Suggested guidelines for the diagnosis and management of urea cycle disorders: First revision. J Inherit Metab Dis. 2019;42: 1192–1230.
- Protocolo de hiperamonemia. Revisión 2021. Irene Martínez de Albéniz Margalef1, Lidia Martínez Sánchez2, Àngels Garcia-Cazorla3, Miquel Villaronga Flaqué4, Lluïsa Hernández Platero5, Aida Ormazabal Herrero6, Camila García-Volpe7. 1Servicio de Pediatría; 2Área de Urgencias; 3Servicio de Neurología; 4Servicio de Farmacia; 5Unidad de Cuidados intensivos pediátricos; 6Servicio de Laboratorio; 7Servicio de Gastroenterología, Hepatología y Nutrición
Más información en Guía Metabólica
- Defectos del ciclo de la urea
- Aciduria propiónica
- Aciduria metilmalónica
- Defectos de la β-oxidación
- Deficiencia de citrina
- Deficiencia de piruvato carboxilasa (PC)
- Mutaciones en el gen GOT2 causan una encefalopatía relacionada con la lanzadera malato-aspartato
- TANGO2: una enfermedad de tráfico de proteínas entre orgánulos
- 16350 lecturas
También te puede interesar

