Deficiencia del transportador de monocarboxilatos tipo 1 (MCT1) como origen genético de cetoacidosis recurrente
Resumen de la cápsula metabólica presentada por la Dra Àngels García-Cazorla en la Unidad de Enfermedades Metabólicas del Hospital Sant Joan de Déu Barcelona el 15 de mayo de 2020.
 como origen genético de cetoacidosis recurrente")
Los transportadores son proteínas cuya función es transportar ciertas moléculas o substancias a través de la membrana plasmática (a modo de lanzadera) para que puedan metabolizarse adecuadamente en el organismo.
Un transportador de monocarboxilatos (MCT) transporta compuestos, que contienen un grupo carboxilo (R-COOH).
¿Qué función tiene el MCT1?
Existen 16 tipos de transportadores de monocarboxilatos, que son específicos para diferentes substancias y tejidos según las especies. El de tipo 1 (MCT1) es un transportador de monocarboxilatos ubicuo (que se halla en muchos tejidos: cerebro, músculo, ojo, testículos, etc) y funciona acoplado a protones. Está codificado por el gen SLC16A1 en humanos.
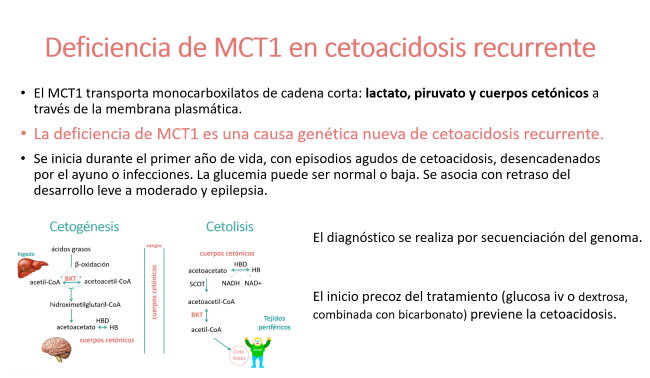
Transporta monocarboxilatos de cadena corta, tales como el lactato, piruvato y cuerpos cetónicos a través de la membrana plasmática.
Permite el equilibrio energético entre los distintos tejidos con diferente capacidad para producir estos compuestos y la regulación (homeostasis) del pH. En el cerebro transporta lactato de los astrocitos a las neuronas.
¿Qué ocurre si se produce una deficiencia de MCT1?
Una deficiencia de MCT1 puede producirse cuando existen mutaciones (cambios estables y hereditarios) en el gen SLC16A1. Se han descrito mutaciones en dicho gen en pacientes con cetoacidosis recurrente.
¿Qué es cetoacidosis?
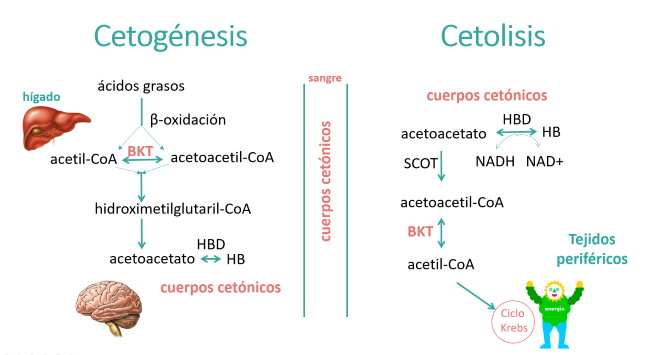
La cetoacidosis se produce cuando hay un desequilibrio entre la cetogénesis o formación de cuerpos cetónicos (3-hidroxibutirato y acetoacetato) y su utilización, llamada cetolisis.
Los cuerpos cetónicos se producen en el hígado mediante la β-oxidación de los ácidos grasos y tienen una importante función en el metabolismo energético.
La cetoacidosis se produce cuando la cetogénesis es mayor que la cetolisis, por defectos de la cetolisis. En estos casos, los niveles de glucosa son normales o bajos, a diferencia de la cetoacidosis diabética en la que los niveles de glucosa son elevados.
Pero la cetoacidosis también puede ser debida un defecto de transporte de cuerpos cetónicos por deficiencia del transportador MCT1 debido a mutaciones en el gen SLC16A1.
¿Qué manifestaciones clínicas tiene una deficiencia de MCT1?
Se manifiesta durante el primer año de vida, con episodios agudos de cetoacidosis, desencadenados por el ayuno o infecciones.
Los primeros 7 pacientes fueron descritos en 2014 (1) y posteriormente se han descrito unos pocos pacientes más (2-4). El diagnóstico se realiza por secuenciación del genoma. Los pacientes con mutaciones en homocigosis tienen un fenotipo más grave, de inicio más temprano, cetoacidosis más profunda, con retraso del desarrollo leve a moderado y un aumento de la prevalencia de epilepsia.
Se asocian anomalías del sistema nervioso central, microcefalia, epilepsia y migraña. Se han descrito alteraciones características en la neuroimagen (agenesia del cuerpo calloso, alteraciones de la sustancia blanca y gris, implicación de los ganglios basales y el tálamo).
La frecuencia de la cetoacidosis disminuye con el tiempo, habiéndose resuelto completamente a la edad de 7 años, lo que sugiere la existencia de mecanismos adaptativos.
En todos los pacientes, el tratamiento con glucosa intravenosa o dextrosa, combinada con bicarbonato da lugar a una rápida desaparición de la acidosis metabólica. El inicio precoz del tratamiento previene la cetoacidosis.
La descripción del defecto del transportador de monocarboxilatos tipo 1 como causa de cetoacidosis recurrente es importante ya que puede explicar el origen de dicha alteración metabólica, que quedaba sin diagnóstico en estos pacientes, dada la dificultad de la confirmación de dicha deficiencia antes de que se dispusiera de las tecnologías genético-moleculares adecuadas.
- 3523 lecturas
También te puede interesar


