Nuevos fenotipos asociados a la β-oxidación peroxisomal: importancia del estudio de la fisiopatología
Resumen de la Cápsula metabólica presentada por el Dr. Alfonso Oyarzábal, en la Unidad de Enfermedades Metabólicas del Hospital Sant Joan de Déu el 5 de junio de 2020.

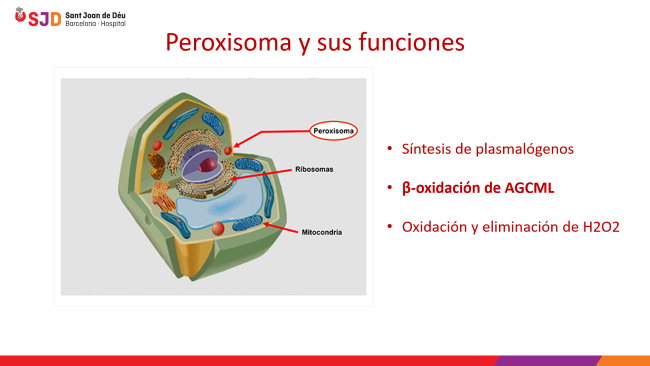
El peroxisoma es un orgánulo que se halla en el citosol celular y contiene diversas enzimas que catalizan muchas reacciones de síntesis y degradación de compuestos, de gran importancia metabólica. El peroxisoma se halla en todos los tejidos, pero predomina en el hígado, en el riñón y en el cerebro durante el período de formación de la mielina (material que recubre las fibras nerviosas y forma la sustancia blanca cerebral).
En Adrenoleucodistrofia ligada al cromosoma X dispones de más información sobre las enfermedades peroxisomales.
¿Cuáles son las funciones del peroxisoma?
Los peroxisomas tienen múltiples funciones, entre las que destacan las relacionadas con el metabolismo lipídico. Entre ellas se hallan las reacciones de degradación, como la β-oxidación de los ácidos grasos de cadena muy larga (AGCML: de más de 22 átomos de carbono), mediante la cual se acorta la longitud de su cadena para que puedan seguir degradándose en el interior de la mitocondria.
Entre las reacciones de síntesis, destaca la formación de plasmalógenos (lípidos complejos localizados en la mielina), colesterol y ácidos biliares.
Otra de las funciones del peroxisoma es la oxidación y eliminación de peróxido de hidrógeno (H2O2), de la cual deriva su nombre (peroxisoma), mediante la enzima catalasa.
¿Qué es la deficiencia de acil-CoA oxidasa 1 (ACOX1)?
La ACOX1 peroxisomal es la primera enzima en la β-oxidación de los AGCML y una gran generadora de H2O2 (especie reactiva de oxígeno tóxica).
La deficiencia de ACOX1 es una enfermedad autosómica recesiva, que causa una rápida y grave pérdida de función del sistema nervioso. Los recién nacidos con deficiencia de ACOX1 muestran hipotonía y convulsiones, posteriormente tienen una pérdida gradual de habilidades de aprendizaje y habla, que generalmente comienza entre las edades de 1 y 3.
A medida que la enfermedad avanza, los niños desarrollan reflejos exagerados, aumento del tono muscular, convulsiones graves y recurrentes y pérdida de visión y audición. Los AGCML están aumentados, porque no pueden ser degradados. La mayoría de los niños con deficiencia de ACOX1 peroxisomal no sobreviven a la primera infancia (1).
Nuevo fenotipo con la mutación (p.N237S) en el gen ACOX1
Recientemente (2), se han identificado tres pacientes con la misma mutación (p.N237S) en ACOX1. Esta mutación no está presente en los padres de ninguna de las tres familias, por lo que es una variante de novo en tres familias no relacionadas.
Los tres pacientes muestran fenotipos similares, incluida una mieloneuropatía (afectación de la médula espinal y los nervios periféricos) progresiva con hipoacusia neurosensorial (sordera), que comienzan entre los 3 y los 12 años (enfermedad de Mitchell).
Los tres pacientes descritos aquí son todos heterocigotos para la variante p.N237S en ACOX1 (enfermedad autosómica dominante).
De hecho, los fenotipos de estos tres casos contrastan mucho con los pacientes con deficiencia de ACOX1 clásica. Además, la variante p.N237S no causa un aumento en AGCML, lo que sugiere un nuevo mecanismo de enfermedad para ACOX1.
¿Cómo se estudia el mecanismo de patogenia en la deficiencia de ACOX1?
Estos estudios se realizan en la mosca Drosophila melanogaster (DM) que también realiza la β-oxidación de AGCML en el peroxisoma. Chung y colaboradores (2) observan que dACOX1 (ACOX1 en la mosca DM) se expresa en las glías (células del sistema nervioso que dan soporte y protección a las neuronas).
La pérdida de función de dACOX1 en DM afecta gravemente a la esperanza de vida, aumenta los AGCML, causa la pérdida de visión y conduce a la pérdida glial y a la reducción de la supervivencia neuronal en la mosca.
Además, Chung y colaboradores demuestran que el aumento de AGCML tiene un efecto neurodegenerativo y, en cambio, la reducción de los niveles elevados de AGCML por otros mecanismos es beneficiosa en la mosca.
¿Cómo funciona la enzima ACOX1 para oxidar los AGCML en el peroxisoma?
Estudios cristalográficos de la proteína ACOX1 demuestran que esta enzima activa es un dímero (dos proteínas unidas). Durante la β-oxidación peroxisomal de AGCML, se genera H2O2, una importante especie reactiva de oxígeno. El H2O2 tóxico se reduce mediante la catalasa peroxisomal, que lo convierte en O2.

¿Cuál es el mecanismo de patogenia de la variante N237S en ACOX1?
A diferencia de las mutaciones causantes de pérdida de función de ACOX1, la nueva variante p.N237S promueve la formación del dímero activo y estabiliza ACOX1, impidiendo su degradación.
Esta variante actúa como una mutación de ganancia de función. Produce niveles elevados de especies reactivas de oxígeno (ROS, radicales libres) en la glía aislada, no altera los niveles de AGCML y conduce a la desaparición de la envoltura de la glía en las moscas DM.
¿Cómo suprimir la producción de especies reactivas de oxígeno tóxicas?
El uso de un potente antioxidante, la N-acetil cisteína amida (NACA), suprime el efecto de estas especies reactivas de oxígeno en la mosca. Podría ser un tratamiento efectivo en los pacientes, pero aún no está aprobado su uso en el hombre.

Resumen
Chung y colaboradores muestran que ACOX1, una enzima peroxisomal necesaria para degradar los AGCML, se expresa principalmente en células gliales. Mutaciones causantes de pérdida de función de ACOX1 provocan una acumulación de AGCML y degeneración glial.
Sin embargo, una mutación en ACOX1 (N237S) causante de ganancia de función identificada en tres pacientes da lugar a un aumento de especies reactivas de oxígeno y pérdida glial, sin aumento de AGCML. Los antioxidantes suprimen su acción de forma potente en la mosca.
La actividad adecuada de la β-oxidación peroxisomal es esencial para la supervivencia glial, y tanto la pérdida como la ganancia de función de la enzima ACOX1 afectan gravemente la función glial en moscas y humanos.
Conclusiones
Mutaciones en un gen pueden causar cambios estructurales en las proteínas con pérdida o ganancia de función. Las mutaciones causantes de pérdida de función (como los defectos enzimáticos o de transporte) son más fácilmente demostrables por sus efectos metabólicos, y por ello, más conocidas.
Distintas mutaciones en un mismo gen pueden causar daño neurológico por diferentes mecanismos fisiológicos. El estudio de la base patofisiológica de casos concretos permite plantear un abordaje terapéutico personalizado más efectivo.
Bibliografía
(1)- Waterman HR, Ferdinandusse S, Wanders RJA. Human disorders of peroxisomal metabolism and biogenesis. Biochemica & Biophysica Acta 2016;1863: 922-933.
- 1876 lecturas
También te puede interesar

 como origen genético de cetoacidosis recurrente")
