Guía clínica para el diagnóstico y tratamiento de los trastornos del ciclo de la urea: Primera revisión (1ª parte)
Häberle J, Burlina A, Chakrapani A, Dixon M, Karall D, Lindner M, Mandel H, Martinelli D, Pintos-Morell G, Santer R, Skouma A, Servais A, Tal G, Rubio V, Huemer M, Dionisi-Vici C. Suggested guidelines for the diagnosis and management of urea cycle disorders: First revision. J Inherit Metab Dis. 2019 Nov;42(6):1192-1230. doi: 10.1002/jimd.12100. Epub 2019 May 15. PMID: 30982989.
")
En 2012, publicamos una guía clínica que resume y evalúa la evidencia de finales de 2011 para el diagnóstico y el tratamiento de los trastornos del ciclo de la urea (DCU). Con una incidencia estimada de 1:35.000-1:69.000, los DCU causan hiperamonemia neonatal (~ 50%) o de inicio tardío, que pueden conducir a discapacidad intelectual o muerte, incluso cuando existen terapias efectivas.
En los 7 años transcurridos desde la publicación de la primera guía, se ha acumulado abundante información novedosa, se ha ampliado la experiencia en el cribado neonatal de algunos DCU, se ha informado de un nuevo trastorno genético causante de hiperamonemia (deficiencia de anhidrasa carbónica Va (CAVA), se ha introducido el fenilbutirato de glicerol como tratamiento, y se han abierto nuevas y prometedoras vías terapéuticas (incluida la terapia génica).
Varios factores, incluido el impacto de la primera edición de estas pautas (leídas y citadas con frecuencia) pueden haber aumentado la conciencia entre los profesionales de la salud y las familias de los pacientes. Sin embargo, el reconocimiento insuficiente y el diagnóstico tardío de los DCU todavía parecen estar muy extendidos.
Por tanto, era necesario revisar las directrices originales para garantizar un marco de referencia actualizado para los profesionales y los pacientes, así como para las campañas de sensibilización. Esto se logró manteniendo el espíritu original de proporcionar un consenso transeuropeo basado en la evidencia sólida (puntuada con la metodología GRADE), involucrando a profesionales en DCU de nueve países en la preparación de este consenso.
Creemos que esta guía, que ha sido revisada por varias sociedades involucradas en el manejo de los DCU, tendrá un impacto positivo en el pronóstico de los pacientes al establecer estándares comunes y difundir y armonizar las buenas prácticas. También puede promover la identificación de vacíos de conocimiento que serán abordados por investigaciones futuras.
1ª versión de la Guía Clínica
Häberle J, Boddaert N, Burlina A, Chakrapani A, Dixon M, Huemer M, Karall D, Martinelli D, Crespo PS, Santer R, Servais A, Valayannopoulos V, Lindner M, Rubio V, Dionisi-Vici C. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis. 2012 May 29;7:32. doi: 10.1186/1750-1172-7-32. PMID: 22642880; PMCID: PMC3488504.
Defectos primarios y secundarios del ciclo de la urea
La presente revisión se centra en los defectos de las cinco enzimopatías clásicas del ciclo de la urea: carbamil-fosfato sintetasa 1 (CPS1), ornitina transcarbamilasa (OTC), argininsuccinato sintetasa (ASS), argininsuccinato liasa (ASL) y arginasa, y también en los defectos secundarios: deficiencias de N-acetilglutamato sintasa (NAGS) y del antiportador mitocondrial de ornitina/citrulina (ORNT1), deficiencia de citrina (citrulinemia tipo 2), la lisinuria con intolerancia a proteínas (LPI), las deficiencias de pirrolina 5-carboxilato sintetasa (P5CS), de ornitina aminotransferasa (OAT) y de anhidrasa carbónica Va (CAVA), que son raras en las poblaciones europeas y aún con evidencia limitada para su manejo (ver Figura).
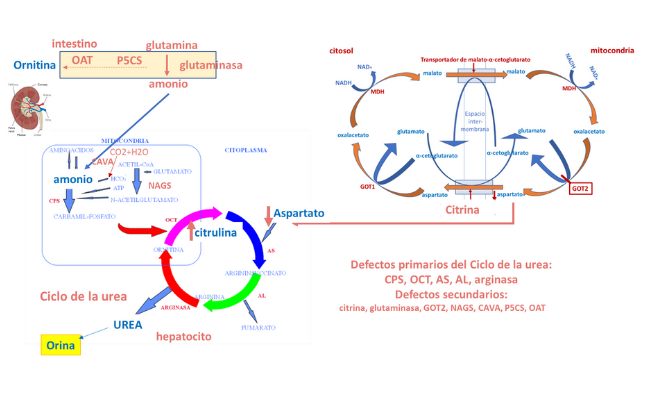
Metodología empleada en las recomendaciones
Para puntuar los niveles de evidencia se adoptó la metodología GRADE (Grading of Recommendations Assessment, Development and Evaluation,): Alto (++++), Moderado (+++), Bajo (++) y Muy bajo (+). Se llegó a un consenso sobre la importancia de los parámetros de resultado (supervivencia, compromiso cognitivo, neurológico y psiquiátrico, afectación hepática, crecimiento y calidad de vida, así como sobre las preguntas clave para conseguir estos resultados.
Objetivo de la guía clínica
El objetivo de esta guía clínica consiste en facilitar y optimizar la toma de decisiones en la atención de un paciente con un defecto del ciclo de la urea (DCU), ya que es un instrumento basado en la evidencia.
Considerando los pacientes individuales, las guías no deben sustituir la toma de decisiones clínicas prudentes, pero pueden proporcionar una base para considerar las opciones diagnósticas y terapéuticas.
Tabla 1.-Signos y síntomas clínicos de presentación aguda y crónica de los DCU
Presentación aguda:
- Nivel alterado de conciencia (desde letargo y somnolencia a coma) similar a encefalitis o intoxicación por drogas.
- Encefalopatía aguda (ver debajo).
- Convulsiones (principalmente en situación de alteración del nivel de conciencia).
- Ataxia: principalmente en situación de nivel alterado de conciencia.
- Episodios similares a un accidente cerebrovascular.
- Pérdida visual transitoria.
- Vómitos y falta progresiva de apetito.
- Insuficiencia hepática, coagulopatía (especialmente en OTCD y HHH)
- Fallo multiorgánico.
- Fallo de la circulación periférica.
- Síntomas psiquiátricos (alucinaciones, paranoia, manía, cambios de personalidad o emocionales).
- “Psicosis posparto”.
- En recién nacidos: cuadro clínico similar a la sepsis, temperatura inestable, distrés respiratorio, hiperventilación.
Presentación crónica:
- Confusión, letargia, mareos.
- Dolores de cabeza, similares a las migrañas, temblor, ataxia, disartria, temblor de aleteo (en adultos).
- Problemas de aprendizaje, discapacidad cognitiva.
- Epilepsia.
- Corea, parálisis cerebral.
- Pérdida visual cortical prolongada.
- Diplejía espástica progresiva o tetraplejia que comienza en la niñez (en ARG1D y síndrome de HHH).
- Aversión a las proteínas, dieta baja en proteínas autoseleccionada.
- Dolor abdominal (recurrente), vómitos.
- Fallo de medro.
- Hepatomegalia, elevación de las enzimas hepáticas.
- Síntomas psiquiátricos: hiperactividad, alteración del estado de ánimo, cambios de comportamiento, agresividad.
- Comportamiento autolesivo.
- Síntomas similares al autismo.
- Cabello frágil (principalmente en ASLD).
- Dermatitis.
- Carácter episódico de signos y síntomas.
- Fenotipo neuropsicológico específico en mujeres heterocigotas de OTCD.
Factores desencadenantes de hiperamonemia en pacientes con DCU:
- Nacimiento del paciente: paso de la vida intrauterina a la extrauterina.
- Infecciones.
- Fiebre.
- Vómitos.
- Hemorragia gastrointestinal o interna.
- Disminución de la ingesta de energía o proteínas (p. ej., ayuno antes de la cirugía, mayor pérdida de peso en recién nacidos).
- Catabolismo e involución del útero durante el período posparto (mujeres OTC).
- Quimioterapia, glucocorticoides en dosis altas.
- Ejercicio físico intenso o prolongado.
- Cirugía con anestesia general.
- Carga de proteínas inusual (p. ej., una barbacoa, nutrición parenteral).
- Fármacos: principalmente valproato y L-asparaginasa / pegaspargasa, topiramato, carbamazepina, fenobarbital, fenitoína, primidona. También se han administrado furosemida, hidroclorotiazida y salicilatos, asociados con la descompensación hiperamonémica.
Resultado: supervivencia
- Pregunta clave: ¿Cómo se puede identificar a los pacientes con un DCU de forma fiable y precoz?
- Recomendación n° 1: Considerar un DCU a cualquier edad, en cualquier deterioro neurológico agudo o intermitente, enfermedad psiquiátrica, insuficiencia hepática aguda, sospecha de intoxicación o en el diagnóstico diferencial de la sepsis neonatal.
- El catabolismo o la sobrecarga de proteínas pueden representar un factor desencadenante.
- Calidad de la evidencia: moderada
Hallazgos de laboratorio
La hiperamonemia es el marcador bioquímico de los DCU, con una concentración de amonio> 500 μmol/L en la mayoría de los pacientes neonatales en el momento de la presentación. Un amonio normal virtualmente excluye un DCU en un recién nacido sintomático (pero no en un paciente mayor).
La medición inmediata de amonio en un entorno de emergencia es crucial, ya que el resultado del paciente se correlaciona con la duración de la hiperamonemia.
Existen dificultades preanalíticas con respecto a la recolección, manipulación, almacenamiento y análisis de muestras de sangre para el análisis de amonio, que causan contaminación y se deben evitar.
Resultado: supervivencia
- Pregunta clave: ¿Cómo se puede identificar a los pacientes con un DCU de forma fiable y precoz?
- Recomendación n° 2: Determinar el amonio en todas las condiciones definidas por la recomendación n° 1 como análisis de emergencia. Ser consciente de las dificultades preanalíticas (posibilidad de contaminaciones).
- Calidad de la evidencia: alta
Resultado: supervivencia
- Pregunta clave: ¿Cómo se puede identificar a los pacientes con un DCU de forma fiable y precoz?
- Recomendación n° 3: Si el amonio está elevado, se deben tomar muestras de sangre para análisis urgente de aminoácidos y acilcarnitinas. A continuación, comenzar con urgencia el tratamiento mientras se esperan los resultados. También debe recogerse orina para análisis de ácidos orgánicos y ácido orótico.
- Calidad de la evidencia: moderada
Resultado: supervivencia
- Pregunta clave: ¿Cómo se puede identificar a los pacientes con un DCU de forma fiable y precoz?
- Recomendación n° 4: Dado que el diagnóstico erróneo más común de los pacientes con un DCU de inicio temprano es la sepsis neonatal, recomendamos considerar la posibilidad de un DCU en el diagnóstico diferencial.
- Calidad de la evidencia: moderada
Análisis bioquímico y enzimático
Los procedimientos clínicos y analíticos estándar generalmente pueden diferenciar entre hiperamonemia debida a errores innatos o debido a otras condiciones.
El siguiente algoritmo para el diagnóstico diferencial de la hiperamonemia se basa principalmente en análisis de diferentes parámetros en plasma/orina.
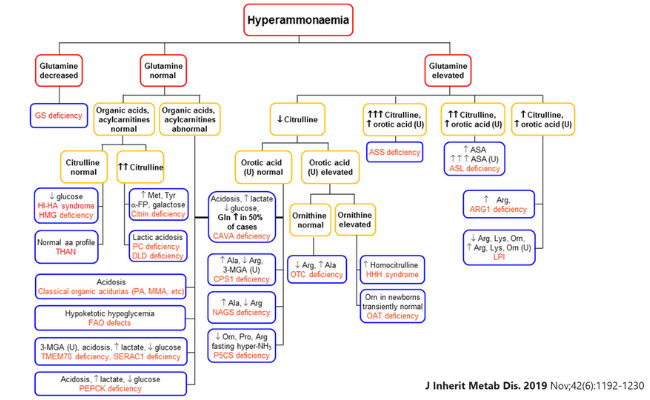
Análisis genético molecular
Se han descrito mutaciones en muchos pacientes para todos los genes de las enzimas del ciclo de la urea y los genes de citrina (SLC25A13) y el transportador de ornitina (SLC25A15) implicados en la citrulinemia II y el síndrome de HHH, respectivamente,
ADN (de células sanguíneas periféricas, tejidos, células cultivadas, o sangre seca) es la muestra preferida. El ARN se utiliza con frecuencia para el análisis de mutaciones de CPS1 dado el número de exones de este gen. El ARN de tejido hepático puede ser útil en OTCD y otros DCU cuando el análisis de ADN de rutina es negativo. El análisis de mutaciones debe incluir investigaciones de dominios regulatorios conocidos.
Resultado: supervivencia
- Preguntas clave: ¿Cómo se puede identificar a los pacientes con un DCU de manera fiable y temprana?
- Recomendación n° 5: Recomendamos realizar pruebas genéticas en los DCU para confirmar el diagnóstico, permitir el consejo genético y, en algunos casos, proporcionar información sobre el curso de la enfermedad. Recomendamos preservar el ADN, los fibroblastos, y/o tejido hepático congelado de pacientes fallecidos con sospecha de DCU.
- Calidad de la evidencia: moderada
Diagnóstico prenatal
Las pruebas prenatales requieren el diagnóstico confirmado en un paciente índice.
Resultado: supervivencia
- Pregunta clave: ¿Cómo se puede identificar a los pacientes con un DCU de forma fiable y precoz?
- Recomendación n° 6: recomendamos el análisis genético molecular como el método de diagnóstico prenatal preferido para todos los DCU.
- Calidad de la evidencia: alta
Tabla 2.-Pruebas prenatales en los DCU: análisis recomendados y requisitos de muestra
- NAGSD: Análisis de mutaciones de ADN en muestras de vellosidades coriales (VC) o líquido amniótico (LA)
- CPS1D: Análisis de mutaciones de ADN en VC o LA. Análisis enzimático, biopsia hepática fetal tardía.
- OTCD: Análisis de mutación de ADN en VC o LA. Análisis enzimático, biopsia hepática fetal tardía.
- ASSD: Análisis de mutación de ADN en VC o LA. Citrulina en LA. Análisis enzimático en VC intactas o cultivadas o LA cultivado.
- ASLD: Análisis de mutación de ADN en VC o LA. Argininosuccinato y sus anhídridos en LA. Análisis enzimático en VC intactas o cultivadas o LA cultivado.
- ARG1D: Análisis de mutación de ADN en VC o LA. Análisis enzimático en eritrocitos de sangre fetal.
- Síndrome de HHH: Análisis de mutación de ADN en VC o LA. Ensayo de función en VC o LA cultivado.
Detección neonatal
Resultado: supervivencia
- Pregunta clave: ¿Cómo se puede identificar a los pacientes con un DCU de forma fiable y precoz?
- Recomendación n° 7: Los pacientes con un DCU se pueden beneficiar de un diagnóstico precoz, por lo que es deseable un cribaje neonatal fiable. Recomendamos considerar la detección neonatal para ASSD y ASLD. En la actualidad, no hay datos suficientes para recomendar la detección neonatal para NAGSD, CPS1D, OTCD, ARG1D y síndrome de HHH.
- Calidad de la evidencia: moderada
Tratamiento de la hiperamonemia aguda
Dado que el pronóstico está muy influenciado por la duración del coma en el momento de la presentación y los niveles máximos de amonio, no se debe retrasar el tratamiento. Los hospitales siempre deberían tener disponibles los medicamentos de primera línea y protocolos escritos basados en consenso sobre cómo proceder.
Las primeras acciones deben ser:
- Detener la ingesta de proteínas.
- Iniciar la glucosa intravenosa (IV) con electrolitos adecuados (Na +, K +)
- Iniciar los medicamentos de primera línea como se describe en la Tabla 3.
- Recogida de plasma y orina con fines de diagnóstico, sin posponer el inicio del tratamiento.
- Transferir al paciente con una crisis hiperamonémica a un centro especializado sin demora.
Resultados: supervivencia, función cognitiva
- Preguntas clave: ¿Cómo se puede mejorar la supervivencia de un episodio de hiperamonemia? ¿Cómo se puede preservar la función cognitiva?
- Recomendación n° 8: Recomendamos iniciar inmediatamente medidas para revertir el catabolismo de proteínas endógenas y para promover la desintoxicación de amonio.
- Calidad de la evidencia: moderada
Antes del tratamiento de la hiperamonemia aguda, es necesario considerar el pronóstico con respecto al resultado del desarrollo neurológico ya que puede influir en la decisión de continuar el tratamiento específico o iniciar cuidados paliativos. El pronóstico se considera muy malo en pacientes con cualquiera de las siguientes características:
- Coma > 3 días
- Presión intracraneal significativamente elevada.
- La concentración de amonio en plasma > 1000 μmol/L generalmente se correlaciona con un pronóstico menos favorable. Sin embargo, concentraciones muy altas de amonio no son un criterio absoluto, siempre deben ser evaluados en asociación con la situación clínica y la duración de la hiperamonemia. Se han descrito pacientes con un resultado normal a pesar de un amonio inicial > 1000 μmol/L.
Fármacos usados en las descompensaciones agudas en DCU
Los captadores de nitrógeno benzoato y fenilacetato son los fármacos de apoyo para tratar el ciclo de la urea. El benzoato se combina con glicina para formar hipurato y la glutamina se convierte en fenilacetilglutamina por acetato de fenilo o su profármaco fenilbutirato. Ambos metabolitos conjugados se excretan en la orina.
La administración de arginina y/o citrulina tiene como objeto maximizar la excreción de amonio a través del ciclo de la urea. El N-carbamilglutamato reemplaza al activador N-acetilglutamato de CPS1.
Comentarios a la Tabla 3:
- Detener las proteínas durante un máximo de 24 h
- Evite las exanguinotransfusiones como causa del catabolismo
- La hiperglucemia puede ser extremadamente peligrosa (hiperosmolaridad)
- Si se produce una hiperglucemia importante con alto nivel de lactato (> 3 mmol/L) reducir velocidad de infusión de glucosa en lugar de aumentar la insulina
- Evitar las soluciones hipotónicas
- Agregar sodio y potasio según los resultados de electrolitos
- Considerar la ingesta de sodio si se utilizan benzoato sódico o PBA sódico
- L-arginina no debe administrarse en deficiencia de arginasa.
- Evite los bolos repetitivos de medicamentos
- Controle los niveles de fosfato y suplementar temprano especialmente durante hemodiálisis.
| Amonio (µmol/L) | Pacientes sin diagnóstico | Pacientes diagnosticados |
| Aumentado por encima del rango normal | Suprimir ingesta proteica | Suprimir ingesta proteica |
|
Administrar glucosa IV en una dosis adecuada para prevenir el catabolismo (10 mg/kg/min en neonatos, 8 mg/kg/min en lactantes, y 6 mg/kg/min en todos los demás) ± insulina |
Administrar glucosa IV en una dosis adecuada para prevenir el catabolismo (10 mg/kg/min en neonatos, 8 mg/kg/min en lactantes, y 6 mg/kg/min en todos los demás) ± insulina |
|
| Monitorizar niveles de amonio cada 3 horas | Monitorizar niveles de amonio cada 3 horas | |
| Amonio >100 y <250 | Iniciar el tratamiento con L-arginina IV y benzoato sódico |
Continuar tratamiento con L-arginina (y/ o agregar L-citrulina para DCU mitocondriales) y benzoato de sodio ± sodio PBA/ fenilacetato, aumentar la dosis o IV |
| Iniciar carbamilglutamato, carnitina, vitamina B12, biotina | Considerar energía sin proteínas (polímeros de glucosa y emulsiones de lípidos) por sonda NG a menos que el niño esté vomitando. | |
| Amonio >250 y <500 | Como anteriormente | Como anteriormente, pero IV |
|
Preparar hemofiltración si encefalopatía significativa y/o nivel de amonio en sangre alto o inicio muy temprano de la enfermedad (día 1 o 2) |
Preparar hemofiltración si encefalopatía significativa y/o nivel de amonio en sangre alto o inicio muy temprano de la enfermedad (día 1 o 2) |
|
|
Iniciar hemofiltración si no hay caída de amonio en 3-6 h |
Iniciar hemofiltración si no hay caída de amonio en 3-6 h |
|
| Amonio > Amonio >250 y <500500 y <1000 | Como anteriormente | Como anteriormente |
| Comenzar hemofiltración inmediatamente | Comenzar hemofiltración inmediatamente | |
| Amonio >1000 |
Evaluar si continuar tratamiento específico o iniciar cuidados paliativos |
Evaluar si continuar tratamiento específico o iniciar cuidados paliativos |
Las dosis de fármacos que se utilizan en la hiperamonemia aguda y las descompensaciones agudas de DCU están detalladas en la publicación: J Inherit Metab Dis. 2019 Nov;42(6):1192-1230.
Detoxificación extracorpórea
En neonatos y niños, la respuesta al tratamiento de emergencia debe evaluarse después de 4 horas.
En adultos, la hemodiálisis o hemofiltración venosa es el tratamiento de primera línea en casos de descompensaciones agudas.
Resultados: supervivencia, función cognitiva, afectación neurológica
Preguntas clave: ¿Cómo puede mejorar la supervivencia el uso de la detoxificación extracorpórea? ¿Cómo se puede preservar la función cognitiva? ¿Cómo se puede prevenir la afectación neurológica?
Recomendación nº 9: En paralelo con tratamiento médico, recomendamos preparar la desintoxicación extracorpórea en pacientes con síntomas neurológicos graves inducidos por hiperamonemia. La desintoxicación extracorpórea debe iniciarse lo antes posible, a menos que el tratamiento médico inicial ya haya dado lugar a una mejora suficiente de los niveles de amonio y de la situación clínica.
Calidad de la evidencia: moderada
Resultados: supervivencia, función cognitiva, afectación neurológica
Preguntas clave: ¿Se puede mejorar la supervivencia en adultos por el uso de detoxificación extracorpórea? ¿Cómo se puede preservar la función cognitiva? ¿Cómo prevenir enfermedades neurológicas?
Recomendación n° 10: Recomendamos que la desintoxicación extracorpórea se considere un tratamiento de primera línea en las descompensaciones hiperamonémicas agudas en adultos.
Calidad de la evidencia: baja
Resultados: supervivencia, función cognitiva, afectación neurológica
Preguntas clave: ¿Puede mejorar la supervivencia el uso de detoxificación extracorpórea? ¿Cómo se puede preservar la función cognitiva? ¿Cómo se pueden prevenir las enfermedades neurológicas?
Recomendación n° 11: Recomendamos hemodiálisis veno-venosa continua o hemodiafiltración como método de elección para la detoxificación del amonio en recién nacidos. Recomendamos considerar diálisis peritoneal, que es menos eficaz para la extracción del amonio, como técnica puente cuando no se dispone de hemodiálisis y para la transferencia de pacientes a un centro metabólico. Recomendamos encarecidamente no realizar exanguinotransfusión para tratar la hiperamonemia.
Calidad de la evidencia: alta
Manejo dietético de la descompensación aguda
Resultados: supervivencia, estabilidad metabólica
Preguntas clave: ¿Cuáles son las intervenciones dietéticas apropiadas en la hiperamonemia aguda? ¿Cómo podemos mejorar la estabilidad metabólica?
Recomendación n° 12: Para el tratamiento de la hiperamonemia aguda, recomendamos establecer y mantener el anabolismo, proporcionando glucosa ± insulina en dosis altas con los electrolitos apropiados (Na +, K +). Los lípidos deben agregarse tan pronto como se hayan excluido los trastornos de oxidación. Recomendamos que la nutrición sin proteínas no exceda las 24 (−48) horas.
Calidad de la evidencia: moderada
Continuación en la 2ª parte de la Guía clínica para el diagnóstico y tratamiento de los trastornos del ciclo de la urea.
Más información en Guía Metabólica
- 3457 lecturas
También te puede interesar


")